Confined migration induces non-lethal DNA damage in developing neurons
Abstract
Migratory cells tend to have soft nuclei that deform and penetrate narrow spaces1,2. Extensive nuclear deformation during migration can cause nuclear-envelope rupture and DNA damage in cancer cells, which may contribute to malignant transformation during tumour progression3,4,5,6. However, the importance of DNA damage in physiological migration is less well understood. Here we demonstrate that the migration of neurons in developing cerebral and cerebellar cortices is accompanied by massive DNA double-stranded breaks (DSBs) due to mechanostress during passage through narrow interstitial spaces. In contrast to many other migratory cells, these DSBs occur without detectable nuclear envelope rupture. Confined migration increases topoisomerase-IIβ covalently bound DSBs, and these lesions are repaired through non-homologous end-joining during brain development without causing cell death. Genome sequencing revealed that DSBs tend to occur at transcriptionally inactive regions. The deletion of ligase IV at the onset of neuronal migration leads to persistent DSB accumulation in cerebellar neurons with moderate transcriptional changes in genes related to synaptic function, neuronal development and stress and immune responses. The mutant mouse develops mild motor deficits in later life, suggesting that the DNA damage generated during normal brain development poses a potential disease risk if left unrepaired.
Main
Owing to their limited regenerative ability, maintaining genome stability in neurons is crucial for preserving brain function throughout the lifespan7,8. However, neurons are continuously exposed to various sources of DNA damage, including intrinsic factors such as oxidative stress, transcription and neural activity, as well as extrinsic factors such as radiation and environmental toxins9,10. Neurons are therefore equipped with robust mechanisms to prevent and correct DNA lesions, as excessive or unresolved damage can contribute to brain ageing and neurodegeneration8,11,12.
During brain development, newborn neurons migrate from their birthplace in the germinal layers to their final destinations in the emerging cortices and nuclei, where they are integrated into functional neural circuits. Migrating neurons squeeze the nucleus, their largest cargo, through narrow spaces crowded with many other cells and extracellular components13,14. Neurons express very low levels of lamin A, which confers high nuclear deformability and migratory ability1,15, but may also render them vulnerable to mechanical stress and resulting DNA damage2,16. In cancer cells and immune cells, severe nuclear deformation during confined migration can cause transient nuclear envelope (NE) rupture and subsequent DNA damage, which has been implicated in cellular senescence and malignant transformation3,4,5,6. By contrast, the impact of nuclear deformation during neuronal migration has not been described. In this study, we investigated whether confined migration influences genome integrity in postmitotic neurons during normal brain development.
Confined migration induces DSBs in neurons
Cerebellar granule neurons (CGNs) are generated by granule neuron precursors (CGPs) in the outer external granule layer (EGL) in the cerebellar cortex (Fig. 1a). Postmitotic CGNs move to the inner EGL and then migrate radially through the molecular layer (ML) to the internal granule layer (IGL) during the first three postnatal weeks in mice. The nuclei of migrating neurons exhibit highly dynamic motion, including frequent rotation and deformation13,17. We first studied DNA damage in the developing cerebellar cortex at postnatal day 6 (P6) using immunofluorescence analysis of γH2AX, a marker for DNA DSBs, along with the cell proliferation marker Ki-67 and the differentiated neuron marker p27Kip1 (Fig. 1a and Extended Data Fig. 1a). Many Ki-67-positive CGP nuclei in the outer EGL were co-stained with γH2AX. Some cells with strong γH2AX signals in the ML and IGL were identified as glial cells on the basis of BLBP expression (Extended Data Fig. 1b). Besides these DSBs, which were presumably generated by replication stress during cell cycle progression, numerous γH2AX foci were observed in postmitotic CGNs in the ML and IGL that were co-stained with p27Kip1 (Fig. 1a and Extended Data Fig. 1a).
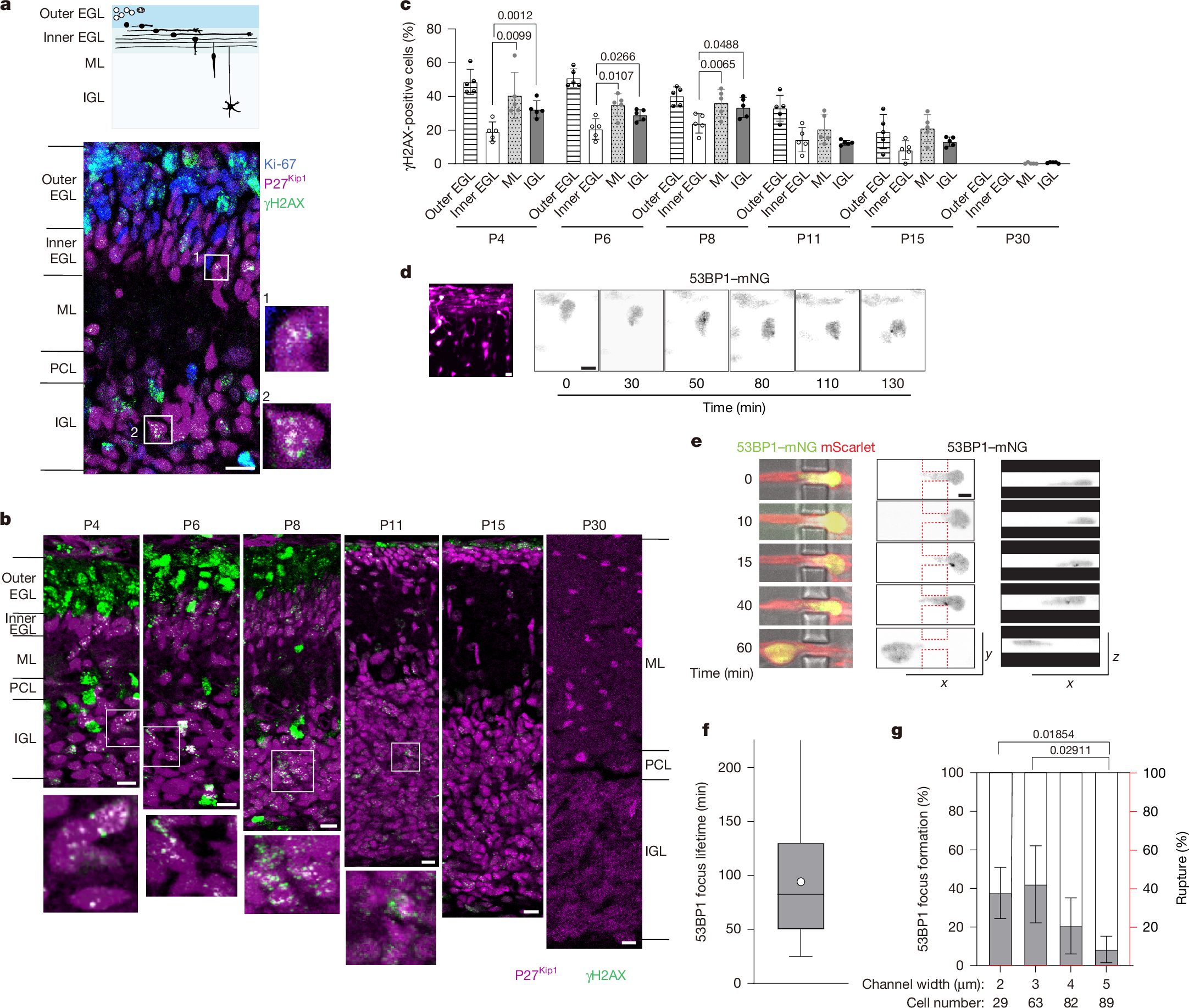
a, Schematic of CGN differentiation during postnatal cerebellar development. CGPs (white) populate the outer EGL. CGNs (black) in the inner EGL extend axons and migrate parallel to the EGL surface and then radially through the ML to reach the IGL (top). Bottom, sagittal section of a P6 mouse cerebellar cortex stained for γH2AX (green), Ki-67 (blue) and p27Kip1 (magenta). Postmitotic CGNs in the boxed regions in the ML and IGL bearing γH2AX foci are magnified on the right. Similar results were obtained from three mice from independent litters. b, The cerebellar cortex from P4 to P30, stained for γH2AX and p27Kip1. The boxed regions in the IGL are magnified at the bottom. c, The percentage of γH2AX-positive cells in the cerebellar cortical layers. Data are mean ± s.d. Two-way repeated-measures analysis of variance (ANOVA) with Dunnett’s multiple-comparison test; P values are shown. The outer EGL was excluded from the comparison. d, Snapshot image of CGNs electroporated with mScarlet (magenta) and 53BP1–mNG (green) in a cerebellar slice from a P7 mouse (left). Right, image sequence of 53BP1–mNG signals in a CGN migrating from the EGL to the ML. See also Extended Data Fig. 1e. e,f, Image sequence of a bright-field view of a CGN transfected with mScarlet and 53BP1–mNG (left) and 53BP1–mNG signals in the xy (middle) and xz (right) planes during migration (e), and the focus life-time in CGNs migrating in 3-µm microchannels (f). The box plots show the median (centre line), 25–75 percentiles (box limits), the whiskers represent the full data range, and the circle indicates the mean of 66 cells analysed. g, The percentage of CGNs that formed new 53BP1 foci (grey) or underwent NE rupture (red) during and/or after passage through channels of the indicated size. Data are mean ± s.d. Statistical analysis was performed using Welch’s ANOVA with Games–Howell multiple-comparison test. Scale bars, 10 µm (a,b), 5 μm (d), 3 μm (e). See also Supplementary Videos 1 and 2. Sample sizes and biological replicates are described in the Source data.
We next examined γH2AX signals at various developmental stages. Abundant γH2AX signals in the outer EGL were observed at P6 and earlier, but sharply declined by P11 as CGPs were exhausted. By contrast, postmitotic CGNs with γH2AX foci in the ML and emerging IGL plateaued from P4 to P8 and persisted until P15 (Fig. 1b). By P30, γH2AX foci were no longer detectable. The proportion of γH2AX-positive neurons was higher in the ML than in the inner EGL at all stages examined, suggesting that postmitotic CGNs acquire de novo DSBs after the onset of radial migration (Fig. 1c). Consistently, γH2AX foci were observed in the nucleus of GFP-positive CGNs migrating in the ML in Neurod1-GFP transgenic mice (Extended Data Fig. 1c).
To further confirm that DSBs are generated during migration, we electroporated the cerebellum with the DSB marker 53BP1-mNeonGreen (53BP1–mNG) and performed live imaging of CGNs in an organotypic cerebellar slice culture. We observed transient formation of 53BP1–mNG foci in CGNs undergoing tangential migration in the inner EGL and radial migration in the ML (Fig. 1d, Extended Data Fig. 1d,e and Supplementary Video 1). To examine whether DNA damage is induced by nuclear deformation in dense neural tissue, we monitored dissociated CGNs cultured on microfabricated substrates with constricted channels18 (1–5 µm width × 5 µm height; Extended Data Fig. 1f). CGNs repetitively passed through channels, undergoing nuclear deformation and compression. We observed transient accumulation of 53BP1–mNG foci during and after passage of the nucleus (Fig. 1e,f and Supplementary Video 2). Migration through the narrower constrictions tended to increase the incidence of focus formation (Fig. 1g and Extended Data Fig. 1f). These results strongly suggest that migrating CGNs undergo DSBs owing to mechanostress while migrating in a confined environment in the developing brain.
We next examined whether DSB formation during neuronal migration is fundamental among neuronal types. Differentiated Purkinje cells migrate from the ventricular zone to the cerebellar cortex primordium at embryonic day 12–18 (E12–18)19. γH2AX foci were detected in the nuclei of postmitotic Purkinje cells labelled by anti-LHX1/5 during and after migration (Extended Data Fig. 2a). Moreover, we observed significant γH2AX foci in the emerging neocortex at E15. The number of γH2AX-positive cells was high in the ventricular zone populated by neural progenitor cells and declined in the intermediate zone as differentiation proceeded. Nuclei with γH2AX foci increased again in the subplate, suggesting de novo DSB formation in postmitotic neurons after the onset of radial migration (Extended Data Fig. 2b). Live imaging of cortical neurons in an organotypic culture also revealed the transient formation of 53BP1–mNG foci during migration from the intermediate zone to the cortical plate (Extended Data Fig. 2c,d and Supplementary Video 3). Together, these observations indicate that at least three types of postmitotic neuron undergo DNA damage during neuronal migration in the developing brain.
Neurons generate DSBs without NE rupture
To investigate the mechanism underlying transient DNA damage in CGNs during confined migration, we adopted a Transwell assay with different pore sizes18. The majority of the CGNs reached the bottom surface within 12 h of seeding through both confined 3-μm pores (56%) and permissive 8-μm pores (79%) (Extended Data Fig. 3a). We observed a significantly higher proportion of cells generating γH2AX foci after confined migration through 3-μm pores compared with cells after non-confined migration through 8-μm pores or unmigrated cells on top of surfaces with 0.4-μm pores (Fig. 2a–c and Extended Data Fig. 3b). Similar results were obtained for 53BP1 staining (Extended Data Fig. 3c,d). We confirmed that γH2AX foci in postmigratory CGNs form characteristic nanodomains in both cerebellar cortex and Transwell cultures, typically clustering around a single spot of Ku70, which is involved in DSB repair through the non-homologous end-joining (NHEJ) pathway20,21 (Extended Data Fig. 3e,f). By contrast, RAD51 foci, indicative of homologous recombination, were not observed in CGNs. More than 20% of CGNs bore multiple γH2AX and/or 53BP1 foci at 6 h and 12 h after transmigration through 3-μm pores, but the levels decreased to the control level at 24 h and thereafter (Fig. 2b,c and Extended Data Fig. 3c,d). To rule out the possibility that the cells were mitotic while migrating and subsequently became postmitotic after reaching the bottom surface, we performed the same assay using CGNs depleted of mitotic CGPs by 24 h preincubation with the cell cycle progression inhibitor palbociclib. A comparable percentage of γH2AX foci was observed, indicating that these DSBs arose from confined migration of CGNs, not from residual replication-induced DSBs originating in CGPs (Extended Data Fig. 3g). A TUNEL assay revealed little to no increase in apoptotic cell death up to 36 h, suggesting that DSBs induced by confined migration are repaired without causing apoptosis (Extended Data Fig. 3h).

a, Immunostaining of CGNs that have migrated through 3-μm or 8-μm pores, 12 h after seeding. b,c, Temporal changes in the percentages of γH2AX-positive CGNs (b; cells with at least five nuclear γH2AX foci) and the average focus number per cell (c) after Transwell migration (3 or 8 μm) and non-migrated nuclei on 0.4-µm pores. d,e, Time-lapse sequences of a HeLa cell (d) and a CGN (e) transfected with mScarlet and mNG-NLS migrating through 3-μm microchannels. See also Supplementary Videos 4 and 5. f, HeLa cells (left) and CGNs (right) were transfected with GFP–cGAS with (3 μm) or without (0.4 μm) Transwell migration (top). Bottom, the percentage of cells with cGAS foci in the nucleus. g, Immunoblot (left) and quantification (right) of TOP2cc formation in migrated (3 μm) and non-migrated (0.4 μm) CGNs at the indicated time after seeding. The two lightest fractions from the CsCl-gradient ultracentrifugation include free TOP2, while the other heavier fractions include TOP2cc. h, Immunoblot (left) and quantification (right) of TOP2cc after 3-µm Transwell migration with or without etoposide (ETO) or epoxomicin. i, γH2AX foci in CGNs after 24 h of 3-μm Transwell migration with or without etoposide. j, Time course analysis of γH2AX-positive nuclei with or without etoposide. k, The average γH2AX focus number (left) and the percentage of γH2AX-postive cells (right) after 3-µm Transwell migration. Each dot represents one independent experiment. Statistical analysis was performed using two-way ANOVA with Tukey’s multiple-comparison test (b, c, h, j and k), two-tailed unpaired t-tests (f) and one-way ANOVA with Tukey’s multiple-comparison test (g); P values are shown. For j, the etoposide-treated and untreated conditions at the same timepoints were compared. Data are mean ± s.d. Scale bars, 5 μm (d–f) and 3 μm (a,i). Sample sizes are described in the Source data.
It has been demonstrated that DSBs induce dynamic structural changes of the NE and promote DSB repair by enhancing the proximity of DNA damage response (DDR) factors associated with the invaginated NE22,23. This raises the possibility that nuclear deformation during confined migration interferes with NE dynamics, thereby delaying the repair of DSBs formed independently of confined migration. We observed that CGNs exhibited NE invaginations after 3 µm confined migration, and these were abolished by treatment with the microtubule depolymerizer nocodazole or by inhibition of the NE protein nesprin-1/2 through overexpression of a dominant-negative nesprin-1 mutant13 (Extended Data Fig. 3i). However, inhibition of NE invagination did not affect the formation or degradation of DSBs (Extended Data Fig. 3j,k). Moreover, ATM, one of the mechanosensitive DDR kinases responsible for NE shape change22, was not required for DSB resolution (Extended Data Fig. 3l). These results suggest that DSB formation and repair during confined migration can occur independently of NE tubule formation.
In some cancer cell lines, DNA damage during confined migration has been shown to be exacerbated by NE rupture, which permits the entry of cytoplasmic nuclease and the leakage of DNA damage repair factors from the nucleus5,6. It has recently been reported that cortical neurons form micronuclei during radial migration, suggesting that neurons also undergo NE damage by nuclear deformation24. We confirmed NE rupture events in HeLa cells using a fluorescent reporter fused to a nuclear localization sequence (mNG–NLS) that rapidly spread in the cytoplasm as cells migrated through constricted channels3,4 (Fig. 2d and Supplementary Video 4). By contrast, in CGNs cultured in the same patterned dish, NE rupture was never observed, even in severely deformed nuclei migrating through narrow pores of 2 or 3 µm width (Figs. 1g and 2e and Supplementary Video 5). HeLa cells expressing catalytically inactive cGAS fused to GFP (GFP–cGAS), a marker of NE rupture3,4,25, showed GFP–cGAS foci in the nuclear periphery after transmigration through 3-μm pores in a Transwell. By contrast, peripheral accumulation of GFP–cGAS was not observed in CGN nuclei with or without confined migration in Transwells (Fig. 2f). NE blebbing and rupture were rare events (that is, less than 3% of CGNs migrating through 3-μm pores), as indicated by immunofluorescence with the nuclear lamina marker lamin B1 and the chromatin marker H4K20me3 (Extended Data Fig. 3l). These findings suggest that, in contrast to many cancer cells, CGNs generate DNA damage by mechanical stress during confined migration independently of NE rupture.
DSBs in neurons involve TOP2
Although NE rupture is a key trigger for DSB formation in many migratory cells, some cell lines have been shown to undergo DSBs in the absence of NE rupture under tight confinement, possibly due to increased torsional strain in the genome during replication6,26. We predicted the involvement of topoisomerase II (TOP2), a key enzyme that relieves torsional stress during replication and transcription, in neuronal DSBs generated in the absence of NE rupture. As previously reported, TOP2β was highly expressed in the nuclei of postmitotic CGNs undergoing radial migration27 (Extended Data Fig. 4a). TOP2 catalyses a temporary DSB in a DNA molecule to allow the passage of another DNA segment, followed by religation. During this process, TOP2 becomes covalently attached to the ends of the DNA break, forming a transient intermediate called the TOP2–DNA cleavage complex (TOP2cc). Failure of TOP2 to religate the break and complete its catalytic cycle results in the accumulation of persistent TOP2ccs, which must then be resolved through proteasomal degradation and DNA repair28. We measured the stalled TOP2ccs in CGNs in a Transwell assay. In unmigrated cells on 0.4-µm-pore filters, we detected free TOP2β unbound to DNA and a very small quantity of TOP2ccs. By contrast, CGNs that migrated through 3-μm pores showed a strong accumulation of TOP2ccs at 8 h, which declined by 12 h (Fig. 2g). This decreas