Analysis of 173,303 exomes and genomes in the Pakistan Genome Resource
Abstract
Naturally occurring loss-of-function variants in human genes enable drug target discovery because they mimic pharmacological inhibition of proteins. However, the study of these genetic variants is constrained by their rarity. Sequencing of diverse populations, particularly those enriched in familial relatedness, has been postulated to promote discovery of rare genetic variants1,2,3. Here we present the Pakistan Genome Resource, a South Asian biobank with high familial relatedness comprising 173,303 participants, who collectively carry naturally occurring homozygous loss-of-function variants in 6,476 genes. We describe the genetic architecture of this population, associations between genes and biomarkers, the distribution of loss-of-function variants across molecular pathways, and recall-by-genotype studies of therapeutically relevant genes. The Pakistan Genome Resource expands the catalogue of human genetic variants, provides a comprehensive genetic reference resource for the Pakistani population, and demonstrates the value of studying diverse cohorts to advance human health.
Main
Analysing the full range of human genetic diversity advances our understanding of disease risk and biological mechanisms, and informs the development of safer and more effective therapies1. Pakistan, like much of South Asia, has been historically under-represented in large-scale endeavours to catalogue genetic diversity. To address this gap, we have established the Pakistan Genome Resource (PGR), a biobank with the primary goal of expanding the breadth of human genetic variation through the sequencing of hundreds of thousands of participants across Pakistan. In doing so, the PGR provides Pakistani communities with comprehensive information about their genetic heritage and its potential relationships to health, and establishes a genomic reference with the potential to improve healthcare outcomes for patients by discovering novel gene–disease associations1,2,3,4.
PGR is a nested case–control cohort comprising whole exomes or genomes of 173,303 individuals recruited from 23 different cities across Pakistan (Fig. 1a). PGR expands on the Pakistan Risk of Myocardial Infarction Study (PROMIS) from 2017 by 15-fold4. With its high levels of familial relatedness, PGR is well-suited to discover instances of genes in which both alleles are identical loss-of-function (LoF) variants. Individuals who are homozygous for LoF alleles have historically been called human gene ‘knockouts’. As this term denotes the purposeful deletion of a gene, we prefer to describe these individuals as carriers of homozygous LoF (homLoF) variants, which is a more accurate description of such naturally occurring genotypes.
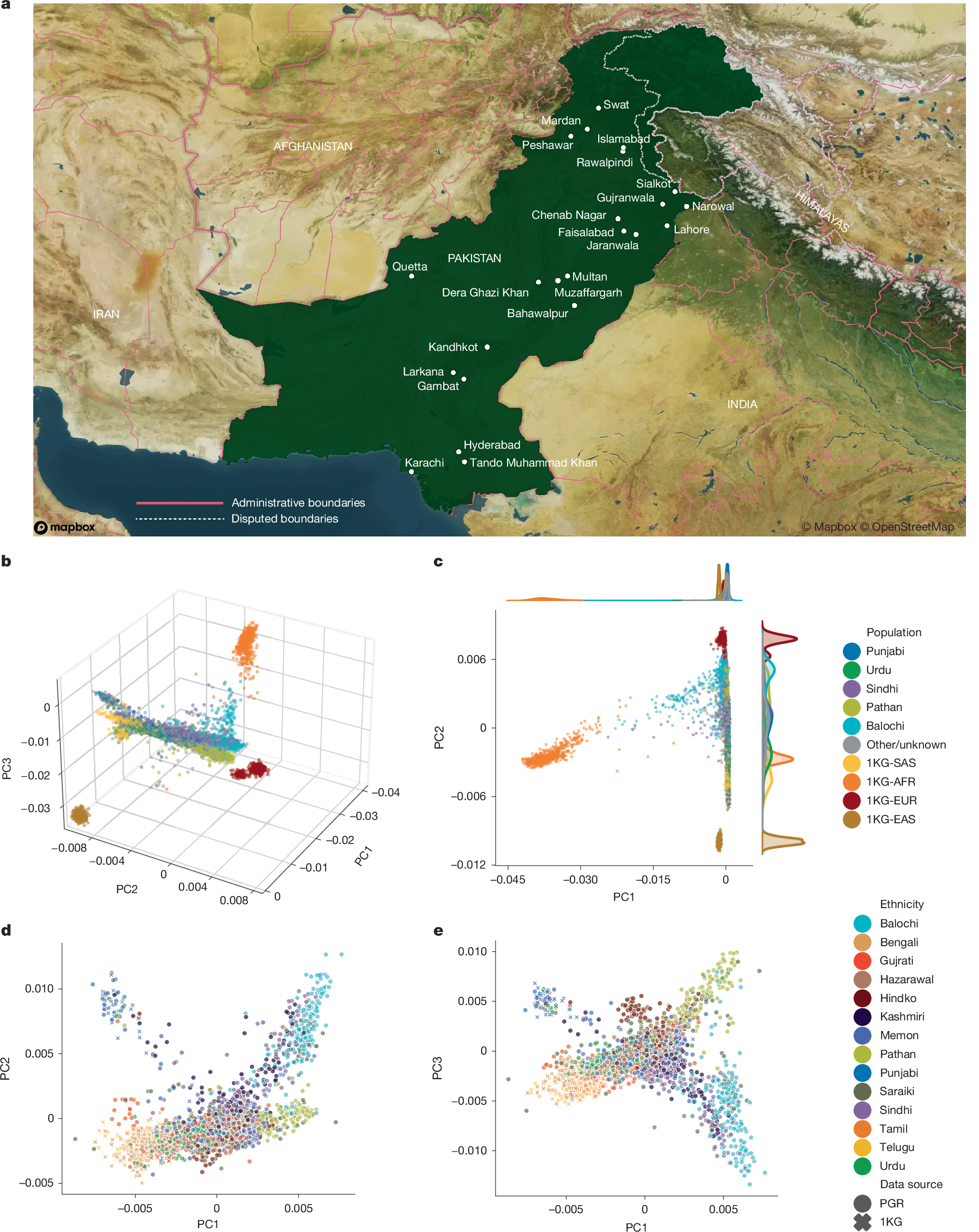
a, Map showing the locations of 23 cities where PGR recruitment centres are located. Copyright OpenStreetMap contributors. b,c, The first three principal components (b) and principal component 1 (PC1) versus PC2 (c) from a PCA of PGR participants and individuals of specific populations from the 1KG dataset: South Asian (SAS; n = 492), East Asian (EAS; n = 515), African (AFR; n = 671) and European (EUR; n = 521). A random selection of 1,000 individuals from each of the 5 largest ethnic groups within the PGR and 1,000 random individuals from the remainder of the PGR were selected from the broader PCA analysis for visualization. d,e, PC1 versus PC2 (d) and PC1 versus PC3 (e) from a separate PCA analysis that is restricted to 1KG SAS and PGR participants with less than 2.5% AFR admixture. A maximum of 200 randomly selected individuals from each ethnicity are shown.
Supplementing participant genotype data, PGR includes a compendium of corresponding clinical data (Supplementary Table 1), including medical history, clinical measurements and disease status for most individuals. PGR facilitates bespoke recall-by-genotype (RBG) studies, enabling familial genotype–phenotype analyses. Such studies can expand the discovery of homLoF variant carriers, as demonstrated in PGR for APOC3 (ref. 4), CORIN5, GDF156, SLC30A8 (ref. 7) and CASP18.
With its spectrum of South Asian-enriched genetic variants, high familial relatedness, RBG capabilities and clinical phenotypes, PGR complements UK Biobank9, FinnGen10, All of Us Research Program11 and the Mexico City Prospective Study12. Here we summarize the regional genetic diversity of PGR, the discovery of homLoF variants across one-third of all protein-coding genes, and present examples of genetic associations and RBG analyses that provide insights into gene function, disease biology and therapeutic translation.
Cohort demographics
PGR comprises nested case–control recruitments across multiple diseases from public and private hospital systems in Pakistan (cohort demographics shown in Supplementary Table 1). We sequenced 166,625 exomes and 6,678 genomes and identified 6,625,995 variants in the coding regions of 19,021 genes (Table 1), including 1,976,965 synonymous (42.1% singletons), 4,186,236 missense (47.0% singletons) and 370,128 putative LoF (pLoF; 59.3% singletons) variants. Of the 6,625,995 coding variants, 3,113,192 (47%) were unique to PGR relative to gnomAD v4.1 non-South Asian individuals13 and 1,974,277 (30%) were unique to PGR relative to all gnomAD inclusive of South Asian individuals.
To measure genetic differences between PGR and other populations, we calculated the fixation index (FST) (ref. 14) for pairs of self-reported PGR ethnicities, reference populations from the 1000 Genomes Project (1KG)15, and the Human Genome Diversity Project (HGDP)16,17 (Extended Data Figs. 1 and 2 and Supplementary Table 2). PGR and other South Asian populations were genetically closer to European and Middle Eastern groups than to East Asian or African groups (Supplementary Table 2, Extended Data Fig. 1). Within PGR, all pairs had an FST between 0.0001 (Sindhi and Kashmiri) and 0.0080 (Balochi and Urdu) (Extended Data Fig. 2 and Supplementary Table 2). The Kalash18 of HGDP diverged from other South Asian populations, with a minimum FST of 0.026 (PGR Pathan). Excluding the Kalash, the maximum genetic divergence among South Asian groups was between PGR Balochi of western Pakistan and 1KG Bengali (FST = 0.0160).
Unsupervised admixture analysis provided a complementary view of population structure. Ethnicities in PGR comprised components characteristic of South Asian and Central Asian reference samples (Extended Data Fig. 3), with admixture proportions differing across ethnic groups (Extended Data Fig. 4). Admixture components from European and Middle Eastern reference samples were more prevalent among the Pathan than in other PGR ethnicities. Recent African admixture was present in some Balochi individuals. East Asian admixture was observed in some Saraiki, Hindko and Hazarawal individuals.
Principal component analysis (PCA) was used to quantify the heterogeneity of PGR subpopulations relative to each other and to non-PGR populations from 1KG (Fig. 1b–e). African, East Asian, European and South Asian populations separated into clusters, with South Asian in PGR following a gradient along principal component 2 (PC2) from European to East Asian (Fig. 1b,c). Consistent with the admixture analysis, Pathan and Balochi ethnicities clustered more closely with Europeans than did other PGR ethnicities, and a subset of Balochi individuals clustered more closely with Africans. Among individuals without African admixture, PC1 distinguished PGR ethnicities along a gradient of proximity to Europeans, as described previously19,20 (Fig. 1d,e). At opposite ends of the gradient were South Indian populations, such as Telugu and Tamil of 1KG-SAS, and the populations of western Pakistan, such as the Pathan and Balochi of PGR. PC2 and PC3 identified two additional clusters, predominantly Balochi or Punjabi, consistent with published substructures among the latter population21.
Consistent with rates of familial relatedness in the region, PGR participants had higher levels of autozygosity than 1KG participants, as measured by runs of homozygosity (FROH) (Fig. 2a). In 166,625 whole-exome sequencing (WES) samples of PGR, mean and median FROH were 0.034 and 0.015, respectively, with variability within and across ethnicities (Fig. 2b). FROH varied across ethnicities in PGR and groups in 1KG-SAS (Fig. 2c). The mean FROH in PGR was within expected ranges of first-cousin (FROH = 0.0625) to second-cousin (FROH = 0.016) parental relatedness, consistent with the 30.6% of participants who self-report having parents who are first cousins.
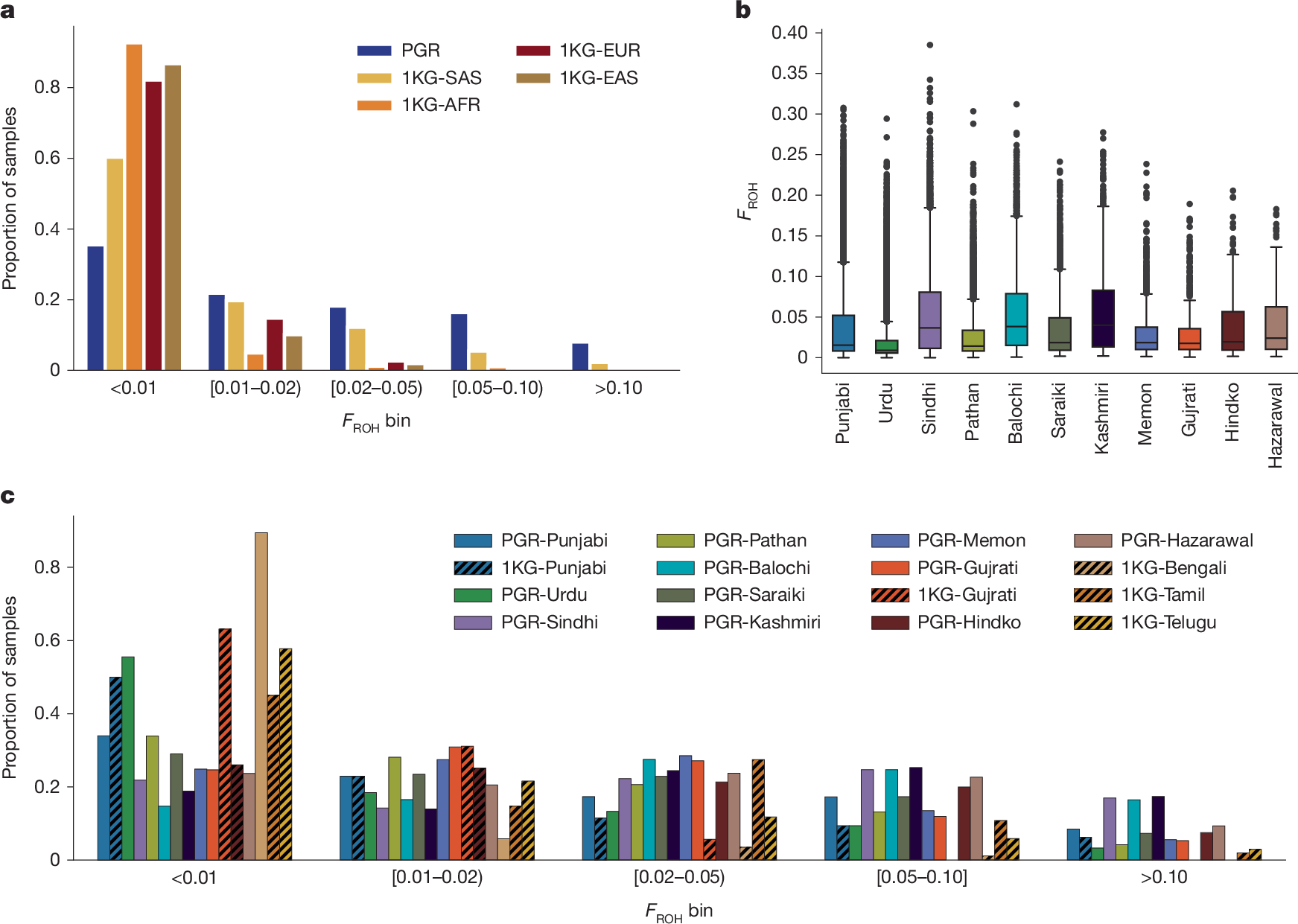
a, Distribution of FROH in PGR for n = 166,625 participants compared to 1KG samples. 1KG analysis sample sizes: SAS, n = 492; EAS, n = 515; AFR, n = 671; EUR, n = 521. b, Box plots showing the distribution of FROH for all self-reported ethnicities within PGR. Boxes show the median FROH and interquartile range (IQR). Whiskers represent 1.5× the IQR. Points are shown for outlier individuals with FROH > 1.5 × IQR. c, Distribution of FROH in PGR ethnicities and 1KG-SAS samples. PGR ethnicity analysis sample sizes: Punjabi, n = 78,392; Urdu, n = 27,295; Sindhi, n = 12,985; Pathan, n = 12,087; Balochi, n = 4,301; Saraiki, n = 3,636; Kashmiri, n = 1,564; Memon, n = 1,111; Gujrati, n = 586; Hindko, n = 441; Hazarawal, n = 278. Analysis sample sizes for 1KG South Asian populations: Punjabi, n = 96; Gujrati, n = 106; Bengali, n = 86; Tamil, n = 102; Telugu, n = 102. Ethnicity-specific subplots exclude individuals for whom ethnicity was not ascertained.
Association analyses
We conducted burden tests to correlate pLoF variants and damaging missense variants to 46 biomarkers (Supplementary Table 3). We filtered high-confidence pLoF variants on the basis of position relative to the end of the transcript, predictions of splicing22, and multi-nucleotide or in-frame ‘rescue’ events, among other filters (Methods). For many genes (including LPA, PLA2G7, APOC3, GPT, ADIPOQ and SELP), pLoF variants (mask1) or pLoF variants combined with damaging missense variants (mask2) associated with reduced circulating levels or activity of the gene products at Bonferroni-adjusted P value thresholds (P < 9.9 × 10−8). Published biomarker associations were confirmed, including associations between triglycerides and pLoF in APOC3 (refs. 4,23) (β = −0.9, P = 8.2 × 10−258); and low-density lipoprotein cholesterol (LDL-C) and PCSK924 (β = −0.9, P = 1.9 × 10−35). These findings validate the pLoF variants, protein measurements and biomarker measurements in the PGR. For comparison, we include associations from the AstraZeneca PheWAS Portal25,26 (Supplementary Table 3).
PGR biomarker data revealed directional causalities that were ambiguous in previous genome-wide association studies. Examples include the genes TIMD4 and HBB. Uncharacterized variants of TIMD4 were previously linked to triglyceride levels27,28. In PGR, TIMD4 pLoF variants were associated with increased triglycerides (β = 0.32, P = 4.7 × 10−10). HBB pLoF variants have previously been linked to reduced haematocrit, increased protection against malaria parasite, increased risk of β-thalassaemia, changes in bilirubin levels and various lipid traits29,30. In PGR, the cumulative allele frequency of HBB pLoF is 0.69% owing to the historical malarial burden in South Asia31. HBB pLoF variants in PGR associated with significantly lower total cholesterol (β = −0.23, P = 1.8 × 10−29), LDL-C (β = −0.26, P = 2.1 × 10−34) and high-density lipoprotein cholesterol (HDL-C) (β = −0.13, P = 1.7 × 10−11); and significantly higher lipoprotein-associated phospholipase A2 (also known as PLA2G7) activity (β = 0.43, P = 8.7 × 10−7) (Supplementary Table 3). In a broader PheWAS, HBB pLoF variants associated nominally with lower levels of apolipoprotein B (ApoB) (β = −0.41, P = 4.0 × 10−6), ApoA1 (β = −0.29, P = 1.0 × 10−3), ApoA2 (β = −0.34, P = 1.2 × 10−4), ApoA5 (β = −0.39, P = 4.0 × 10−5) and ApoC3 (β = −0.25, P = 4.3 × 10−3); higher estimated glomerular filtration rate (eGFR) (β = 0.07, P = 1.1 × 10−5); lower random glucose (β = −0.05, P = 2.3 × 10−2); and lower odds of myocardial infarction in case–control recruitment (odds ratio (OR) = 0.73, P = 1.4 × 10−4) (Supplementary Table 4). Broadening the analysis to myocardial infarctions across all of PGR achieved a more robust association albeit without achieving genome-wide significance (OR = 0.75, P = 1.0 × 10−6). We note that these results highlight candidate associations that still need to be replicated in independent studies.
Having established variant–biomarker associations, we next examined whether increased autozygosity was associated with clinical phenotypes across the cohort. We tested associations between autozygosity (per FROH of 0.0625, the theoretical expectation of FROH for offspring of first-cousin parents) and 16 quantitative and 18 binary phenotypes (Extended Data Fig. 5 and Supplementary Tables 5 and 6). After multiple-testing correction (threshold of P = 1.5 × 10−3), FROH was significantly associated with several traits, including reduced odds of having children (OR = 0.78, P = 4.8 × 10−32), fewer total children (β = −0.07, P = 1.3 × 10−72), fewer years of educational attainment (β = −0.04, P = 2.0 × 10−38) and lower odds of having ever used tobacco (OR = 0.89, P = 3.1 × 10−30). FROH was associated with smaller body size, including body mass (β = −0.08, P = 1.3 × 10−102), height (β = −0.05, P = 1.6 × 10−48) and body mass index (BMI) (β = −0.05, P = 2.5 × 10−35). FROH was associated with a modestly favourable cardiometabolic profile, including significantly lower glucose (β = −0.02, P = 2.3 × 10−6), lower LDL-C (β = −0.03, P = 5.5 × 10−12), total cholesterol (β = −0.04, P = 6.1 × 10−20) and lower systolic (β = −0.02, P = 1.7 × 10−4) and diastolic blood pressure (β = −0.02, P = 5.2 × 10−4). Higher levels of autozygosity were associated with significantly lower odds of myocardial infarction (OR = 0.92, P = 2.1 × 10−8), diabetes mellitus (OR = 0.96, P = 6.8 × 10−5) and hypertension (OR = 0.94, P = 1.8 × 10−10). FROH was associated with significantly higher odds of tuberculosis (OR = 1.12, P = 4. 6× 10−4) and chronic kidney disease (OR = 1.42, P = 2.5 × 10−11). Associations remained consistent after adjusting for educational attainment and tobacco usage as covariates (Extended Data Fig. 5; Supplementary Table 5 and Supplementary Table 6).
To test whether the association between FROH and myocardial infarction was mediated by effects of FROH on other measured covariates of cardiovascular disease, we performed analyses restricted to myocardial infarction cases and controls for whom all of the following parameters were available: BMI, glucose, LDL-C, HDL-C, total cholesterol, triglycerides, tobacco usage, and systolic and diastolic blood pressure. Among these 15,894 individuals with myocardial infarction and 20,229 controls, FROH was associated with lower rates of myocardial infarction (OR = 0.91, P = 6.9 × 10−6). Inclusion of the parameters above as covariates modestly attenuated the effect size (OR = 0.93, P = 1.1 × 10−3).
To explore whether other factors contributed to the associations, we repeated the analyses by self-reported ethnicity, sex, self-reporting of first-cousin parents, and age (Supplementary Tables 5 and 6). Heterogeneity in effect sizes was statistically significant by ethnicity for educational attainment (P = 2.0 × 10−5), number of children (P = 6.4 × 10−8) and HDL-C (P = 3.4 × 10−4), whereas no heterogeneity was observed for the other traits, suggesting that correlates of ethnicity do not confound the remaining associations (Extended Data Fig. 6). No significant differences in associations were observed between male and female participants after multiple-testing correction (Extended Data Fig. 7). Subsetting to individuals with high autozygosity who self-reported to have first-cousin parents32 eliminated the association between autozygosity and educational attainment, suggesting that educational status is confounded by social or economic factors (Extended Data Fig. 8). However, the other associations were unchanged, suggesting that the observed effects are not driven by these socioeconomic confounders. We cannot exclude the possibility that other unmeasured socioeconomic confounders contribute to the observed associations. To test for age dependence, we binned across ages (Extended Data Fig. 9) and observed significant heterogeneity for educational attainment (P = 3.4 × 10−8), number of children (P = 3.2 × 10−5), having had children (P = 6.2 × 10−6), diabetes (P = 9.0 × 10−6) and hypertension (P = 1.9 × 10−8). For example, FROH was associated with increased risk of diabetes (OR = 1.16, P = 9.09 × 10−3) and hypertension (OR = 1.18, P = 1.02 × 10−3) in individuals below 30 years of age and decreased risk of diabetes (OR = 0.94, P = 1.47 × 10−4) and hypertension (OR = 0.90, P = 1.54 × 10−10) in individuals aged 60–69 years, the largest age group in the PGR (Extended Data Fig. 9 and Supplementary Table 6).
homLoF variants
Across all sequenced PGR participants, 6,476 genes were predicted to exhibit homLoF in at least one participant (Fig. 3a and Supplementary Table 7), which represents one-third of all protein-coding genes. Of these 6,476 genes, 3,323 (51%) were uniquely homLoF in PGR relative to 761,616 non-South Asian individuals in gnomAD; 2,298 (35%) were uniquely homLoF in PGR relative to all 807,162 individuals in gnomAD (Fig. 3b and Supplementary Table 8). There were 7,954 genes with homLoF variants in PGR and gnomAD combined (Supplementary Table 8).
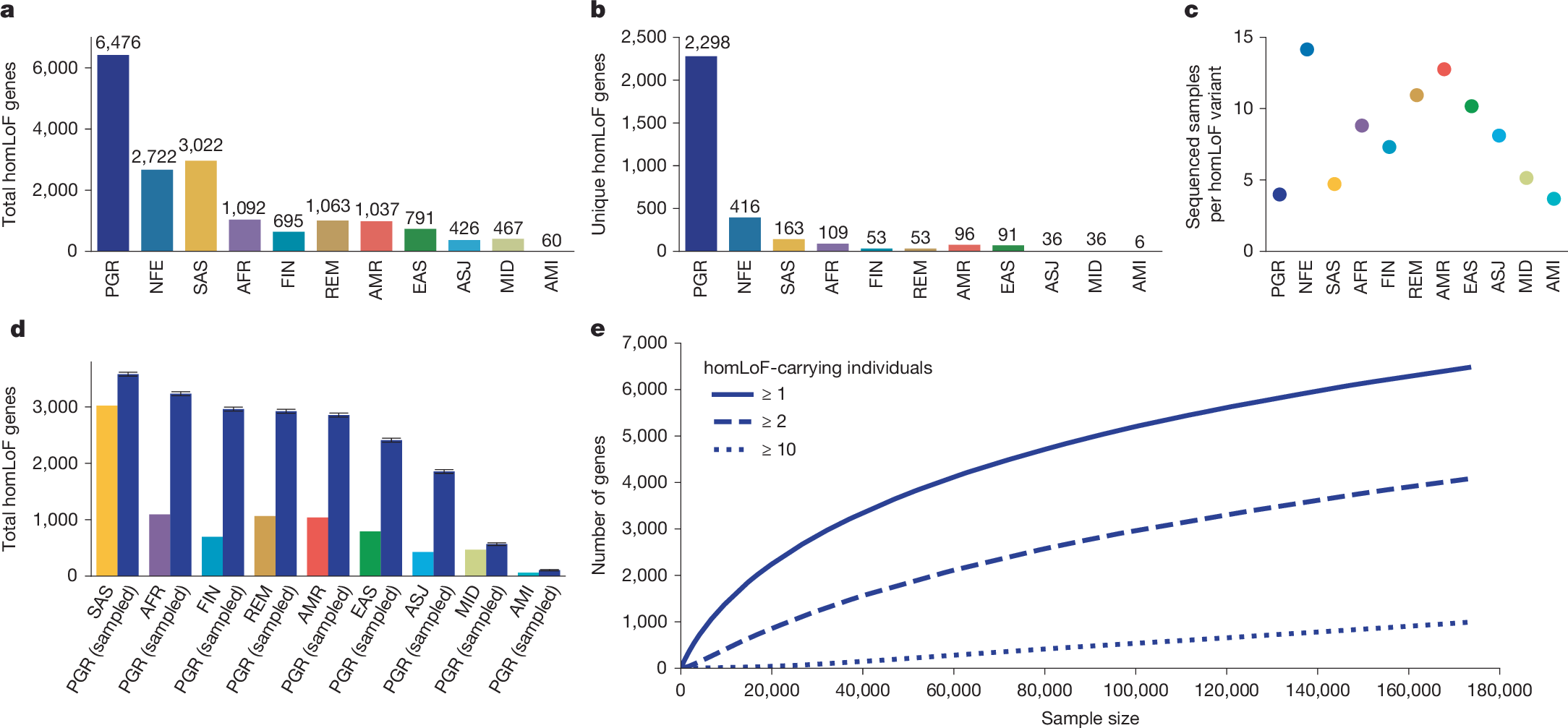
a, The number of genes with at least one homLoF-carrying individual in the PGR and gnomAD v4.1.0 genetic groups. b, The number of genes for which a homLoF variant carrier was found in only one population. c, The number of sequenced samples per homLoF variant observed in PGR and gnomAD v4.1.0 genetic groups d, The numbe