Lethal plague outbreaks in Lake Baikal hunter-gatherers 5,500 years ago
Abstract
Plague is among the most devastating diseases in human history1. However, early strains of the plague-causing bacterium Yersinia pestis lacked virulence factors that are required for the bubonic form until around 3,800 years ago2,3. Consequently, the morbidity and mortality of early plague strains remain unclear. Here we describe early plague strains that are associated with two phases of outbreaks among mid-Holocene hunter-gatherers near Lake Baikal in southeast Siberia, beginning from about 5,500 years ago. These outbreaks occur across four hunter-gatherer cemeteries, with a 39% detection rate for plague infection. By reconstructing kinship pedigrees, we show that small familial groups were affected, consistent with human-to-human spread of disease, and that the first outbreak occurred within a single generation. The infections appear to have resulted in acute mortality, especially among children (aged 8 to 11 years). We further note functional differences, including in the ypm superantigen locus, which is also present in present day Yersinia pseudotuberculosis. The new strains diverge ancestrally to known Y. pestis and constrain the timing of its emergence, indicating that this happened before approximately 5,700 years ago. These findings show that plague outbreaks happened earlier than previously thought and were indeed lethal. We contend that the occurrence of outbreaks among mid-Holocene hunter-gatherer communities well outside the sphere of Late Neolithic Europe challenges the notion that higher population densities and lifestyle changes during the Neolithic agricultural transition were prerequisites for plague epidemics.
Main
The analysis of ancient pathogen genomes has significantly expanded our understanding of the evolutionary history of human infectious diseases (for example, Salmonella enterica4 and hepatitis B5), although this has principally been in the context of farming or pastoralist communities. Y. pestis, the aetiological agent of plague, is perhaps the most studied in this regard, and has had devastating consequences on human populations for millennia. Historical outbreaks of plague account for some of the most fatal events in human history1. The recovery of ancient DNA from plague victims has afforded extraordinary insights into the origins and evolution of plague at the time of these events6,7, and, remarkably, revealed infections in prehistoric individuals across Europe8. Historically and today, plague is associated with transmission via fleas from rodents, which successfully adapted to a human commensal niche in the Neolithic9. Genomic analysis of prehistoric plague indicates that in early diverging strains, key genetic adaptations required for flea-mediated transmission of the disease and bubonic infection are absent2,3, leading to uncertainty over the transmission route and severity of these strains.
The detection of early plague cases across multiple generations of Late Neolithic farmers has been used to link outbreaks of the disease to a prolonged demographic decline between about 5,300–4,900 calibrated years before the present (cal bp)10,11, although an alternative explanation attributes the decline to agricultural crisis12,13. The former interpretation has been controversial, with others suggesting infections as more closely resembling benign foodborne enteritis14. The similarity or otherwise of these early strains to Y. pseudotuberculosis—the closest relative of Y. pestis—has been an important point of interest through such discussions, and based on existing ancient genomes, Y. pestis has been estimated to have diverged from Y. pseudotuberculosis some time in the past 50,000 years (refs. 8,11,15).
Studies of prehistoric plague genomes from Late Neolithic and Bronze Age (LNBA) strains predominantly date to between 4700–2400 cal bp (refs. 3,8,16), and are typically defined as one of two lineages, depending on the presence (LNBA+) or absence (LNBA−) of the ymt gene3. ymt encodes Yersinia murine toxin, which enhances bacterial survival in the flea digestive tract during the transition period between rodent and human hosts, and thereby the flea bite-transmitted bubonic form of plague in humans17. Lineages of Y. pestis that diverged prior to these LNBA clades have also been identified in a handful of Neolithic Swedish individuals (5200-4850 cal bp)10,11 and a Latvian individual with western hunter-gatherer ancestry (5300–5050 cal bp)15. These genomes lack classic virulence genes (YpfΦ prophage and ymt), although pangenomic analysis revealed the presence of the locus encoding for Y. pseudotuberculosis-derived mitogen (YPM), a superantigenic toxin associated with Y. pseudotuberculosis (but not later Y. pestis strains). This raises intriguing questions about the possible severity of early strains of plague; subsequent LNBA− strains show substantial gene loss, although the virulence potential of these are unknown3. Evidence regarding the demographic impact of plague infection on prehistoric populations has so far been lacking in these studies.
Middle Holocene hunter-gatherers around Lake Baikal, southeast Siberia, have been the focus of intensive archaeological study by the Baikal Archaeology Project, yielding important datasets for framing prehistoric hunter-gatherer lifeways18,19. These groups demonstrate remarkable continuity of hunter-gatherer lifeways and subsistence, evidenced by an extensive archaeological record of mortuary sites from between about 8500–3500 cal bp (ref. 20). The genomes of sampled hunter-gatherers indicate a long-term continuum of Ancient North Eurasian and North East Asian ancestry until c. 4500–4000 cal bp (refs. 21,22) (Extended Data Figs. 1 and 2). By this period, cases of plague from human remains corresponding to the LNBA− strain are documented sporadically among Early Bronze Age burials22,23. Zoonotic spillover events causing plague infections in this region remain a major health concern to this day24. These are principally associated with marmots, the primary zoonotic reservoir of plague in the region25,26. To explore health and community structure in prehistoric hunter-gatherer groups, we analysed ancient human and pathogen DNA from four cemetery sites in Cis-Baikal (the lake’s western and northern region) across two separate outbreaks dated to 5520–5265 cal bp and 5315–4235 cal bp (95.4% confidence intervals for modelled date ranges based on individuals with detected plague cases, corrected for freshwater reservoir effects; Supplementary Note 4). The long tail for the second outbreak date range (Fig. 1b) is due to this only consisting of two direct dates, although the highest likelihood date range for this is approximately 5050–4850 cal bp.
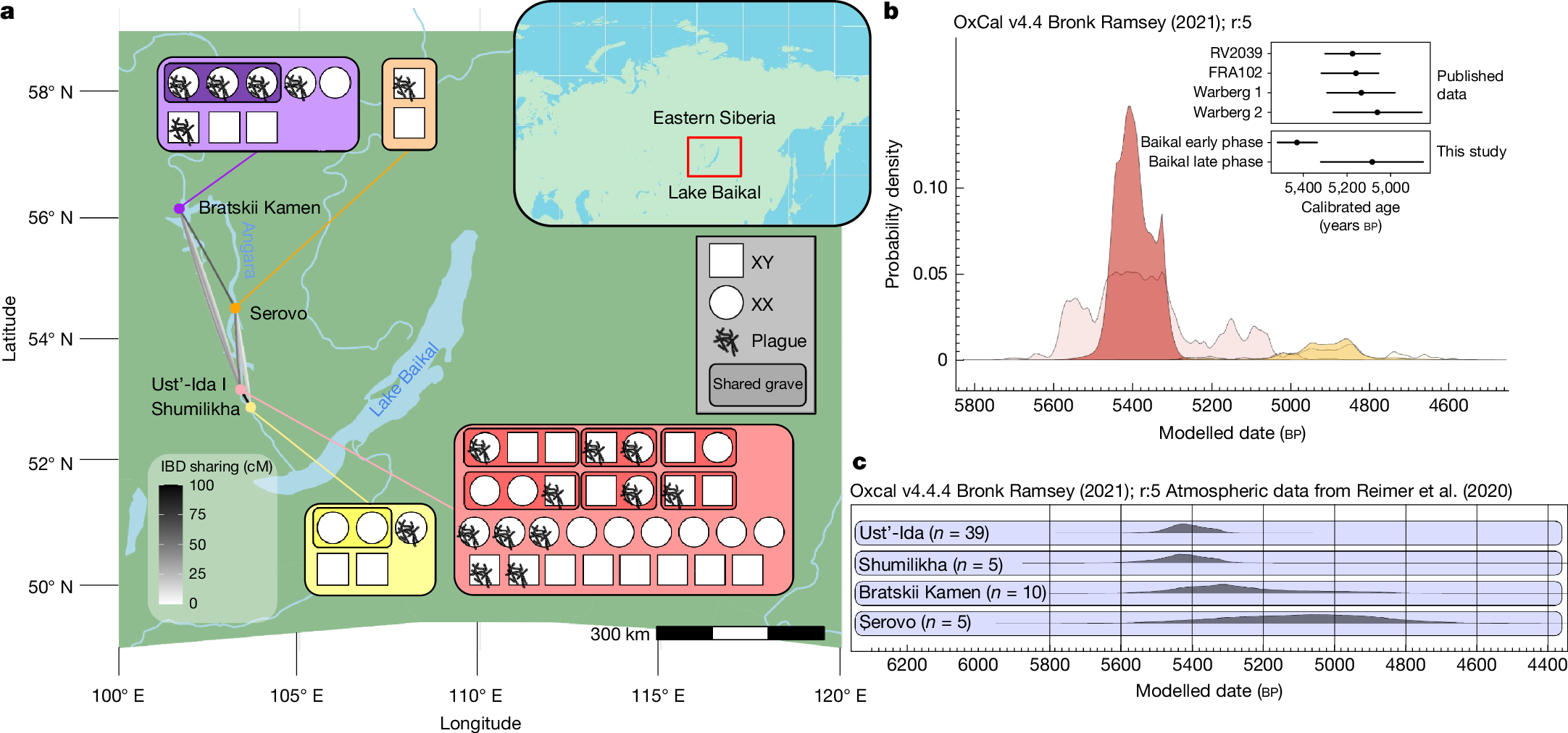
a, Locations of affected cemeteries on the Angara River northwest of Lake Baikal, and IBD sharing between the sampled occupants of cemeteries (pairwise sharing lines between sites; greyscale ramp indicates total IBD sharing in segments larger than 3 centimorgans (cM) totalling more than 10 cM in pairwise relationships between sites; Supplementary Note 2). Inset represent the sampled individuals at each site (31 Ust’-Ida I, 8 Bratskii Kamen, 2 Serovo and 5 Shumilkha), with plague detections indicated, and shared graves indicated by shaded areas around individuals. Maps created using Natural Earth Data. b, Kernel density estimates plotted within Bayesian models of the date ranges for the early (red) and late (dark yellow) phases of plague outbreaks at Baikal. Lighter shaded areas correspond to the summed probability distributions prior to modelling; dates used are from individuals identified with plague only. Inset, 95.4% confidence intervals for modelled date ranges at Baikal are shown compared with those from other pre-LNBA plague cases: RV2039 from Latvia15, Warberg 1 and Warberg 2 from Germany61 and FRA102 from Sweden11. c, Kernel density estimate modelled radiocarbon date distributions for the four cemetery sites, for all radiocarbon-dated post-weaning age humans (or associated deer tooth pendants), irrespective of DNA sampling. Radiocarbon date modelling undertaken with OxCal v.4.4.465,66.
Outbreaks of basal plague strains
We generated shotgun-sequenced ancient DNA from 46 Late Neolithic individuals and examined this data for presence of pathogens (Methods). This revealed a conspicuously high occurrence of Y. pestis among these individuals, more so than any other pathogen. Y. pestis was detected in 18 individuals, indicating 2 distinct phases of outbreaks of plague infection—separated by between 4 and 6 centuries—in 4 cemeteries (Fig. 1a,b). These occur across two phases at Shumilikha, Ust’-Ida I, Bratskii Kamen and Serovo (see Fig. 1c), with cases from Bratskii Kamen in both the first and second phases. These sites are all located on banks of the River Angara, a major watercourse draining from Lake Baikal, with a rich fishery27. Stable carbon and nitrogen isotopic data from individuals at Ust’-Ida I evidence consumption of both local fish and terrestrial game27. Burials at Ust’-Ida I and Shumilikha correspond to the Isakovo mortuary tradition (characterized by bodies that are typically oriented parallel to the river, and the presence of grave goods such as mitre-shaped clay vessels, lithic arrowheads and bone or antler points), whereas those at Bratskii Kamen and Serovo correspond to the Serovo mortuary tradition (with bodies frequently oriented perpendicular to the river; bifaces and egg-shaped pots as grave goods are the main features; see also Supplementary Note 1). At Ust’-Ida I, we also detect reads aligned to the zoonotic pathogen Brucella, the cause of brucellosis, in one individual (#26.04; Supplementary Note 3). The two plague outbreaks are grouped by the predominant burial practices at each cemetery: Isakovo-style graves in the first outbreak and Serovo-style graves in the second phase (Supplementary Note 1), which are contemporaneous at Lake Baikal between around 6000–5000 cal bp (ref. 27). This period is defined locally as the Late Neolithic, following Siberian archaeological terminology, where the Neolithic is defined on technological criteria such as the introduction of the bow and arrow, clay vessels and stone grinding techniques (domestic plants and animals other than dogs are absent), although these communities remain as hunter-gatherers until the encroachment of pastoralism in the Late Bronze Age. Grave sites comprise the vast majority of the Cis-Baikal archaeological record, and designations such as the Late Neolithic are later categorizations, applied to distinct sets of burial characteristics and grave goods that broadly correspond to different periods. All four cemeteries were also used during the Early Neolithic (7650–6660 cal bp) and Early Bronze Age (4970–3470 cal bp)27, although only their Late Neolithic components are considered here.
Pairwise sharing of identity-by-descent (IBD) segments between individuals at these cemeteries indicates recent shared ancestry. Although they are up to 340 km apart, the Angara river would readily have facilitated travel. Very low rates of inbreeding were detected and a high effective population size based on runs of homozygosity was inferred using hapROH (maximum likelihood estimate: 18,219 individuals, 95% confidence interval 9,445–42,062). This is consistent with the scenario of highly mobile, exogamous hunter-gatherer groups.
Within the hunter-gatherer individuals analysed here, the highest number of detected plague infections was at Ust’-Ida I, which is also the largest Isakovo mortuary site in Cis-Baikal. Here, we found a 35% detection rate (11 out of 31 individuals sequenced), including burials #14 and #56.01, for which human genome data were previously reported21. Across other sites, we identify one high-coverage plague genome at Shumilikha, four lower coverage genomes from Bratskii Kamen, and one medium coverage genome from Serovo. Overall, we observe a 39% detection rate across Late Neolithic individuals at these cemeteries (from dental cementum). In comparison, quantitative PCR screening of known Mediaeval plague victims at Smithfield, London, UK28 returned a detection rate of 5.7% from bone and 37% from dental pulp tissue (overall 20%), indicating a high rate of false negative plague detection using ancient DNA. To prevent misrepresentation of data, all ancient individuals with screening data from the affected sites are reported here (human autosomal genome coverage ranges from 0.001× to 1.9×, average 0.65×). Direct radiocarbon dates were obtained from nearly all the individuals within the Late Neolithic components of these cemeteries (a total of 58, including those previously reported from Ust’-Ida I29; Supplementary Data 7).
Y. pestis genomes identified between the two Baikal phases of outbreaks were found to diverge ancestrally to the current known clade of ancient and modern plague strains (Fig. 2). We confidently assign these to Y. pestis from their phylogenetic position, and also the presence of virulence genes and plasmids characteristic of Y. pestis (Extended Data Fig. 3 and Supplementary Note 3). This phylogeny was built using genomes obtained from Shumilikha Burial #34 (6.4× coverage) from the first phase, and from Bratskii Kamen Burial #22 (1.6×) and Serovo Burial #10 (1.0×) from the second phase. Eight lower coverage genomes were phylogenetically placed using UShER30. The UShER algorithm finds the most parsimonious placement on the tree, selecting the node with the greatest number of descendents if multiple are equally parsimonious, and ignores missing genotypes. Placement of all low-coverage genomes at the same basal node is partly due to data missingness, although consistent with the position of the three higher coverage Baikal genomes. Bayesian inference of node dates was undertaken following an approach to account for the effects of recombination within bacterial phylogenies31,32 (Methods and Supplementary Note 3). The emergence of Y. pestis as a clonal species of Y. pseudotuberculosis occurs some time between the divergence of the lineage that gives rise to Y. pestis (labelled node A in Fig. 2) and the most recent common ancestor of available Y. pestis genomes (node B in Fig. 2). The upper bound provided by the former is likely to be substantially affected by the paucity Y. pseudotuberculosis genomes sequences, and could well be more recent if phylogenetically closer serovars were identified. Nonetheless, this lower bound (with a mean date of 5,709 years ago) revises a previous divergence estimate of 4,810–5,122 years ago33, as would be expected by including Y. pestis genomes older than this range (other estimates of this have ranged from 6,000 to 50,000 years ago8 and 7,400 years ago15). The phylogeny supports the conclusion that Y. pestis first evolved from a variant of the O:1 Y. pseudotuberculosis strain (represented by a genome from serotype O:1c, European Nucleotide Archive (ENA) accession: SAMEA7160327), consistent with previous findings34 reporting the inactivation of the O-antigen gene cluster as a step towards the evolution of Y. pestis. Between the two phases, we observe small genetic differences between strains in distinct private mutations in the first and second phase strains (with strict filters for genotype calling; Methods and Supplementary Note 3); this is also clear from the position of nodes in Fig. 2. Although mutation rates in Y. pestis are known to be highly variable within different lineages33, this result is consistent with a scenario of related strains resulting from separate zoonotic spillover events from a local animal reservoir.
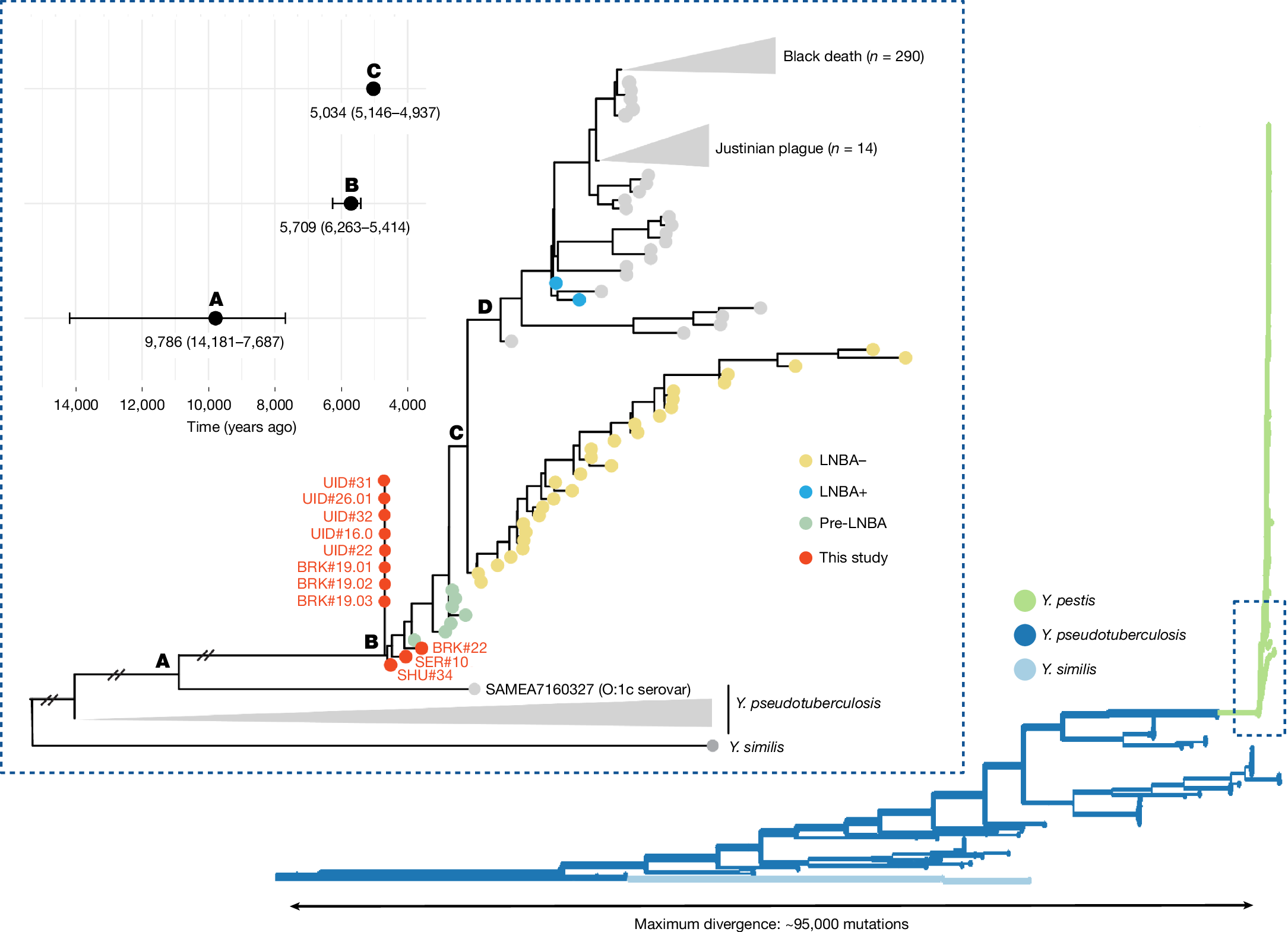
Right, the overall topology of the Y. pseudotuberculosis species complex is shown from a mutation-annotated tree based on 448 genomes (branch lengths indicate distance by mutations). Inset, a simplified version of this phylogeny, with prehistoric plague strains shown in particular (some branch lengths truncated). The three Baikal