αKG-mediated carnitine synthesis drives DNA repair via histone acetylation
Abstract
Homologous recombination (HR) deficiency increases sensitivity to DNA-damaging agents that are commonly used to treat cancer1. In HR-proficient cancers, the metabolic mechanisms that drive response or resistance to DNA-damaging agents remain unclear. Here we have identified that depletion of α-ketoglutarate (αKG) sensitizes HR-proficient cells to DNA-damaging agents by metabolic regulation of histone acetylation. αKG is required for the activity of αKG-dependent dioxygenases2 (αKGDDs), and previous work has focused almost exclusively on the demethylase functions of αKGDD. Using a targeted CRISPR knockout library consisting of 64 αKGDDs, we discovered that trimethyllysine hydroxylase epsilon (TMLHE), the first and rate-limiting enzyme in de novo carnitine synthesis, is necessary for the survival of HR-proficient cells in the presence of DNA-damaging agents. Unexpectedly, αKG-mediated TMLHE-dependent carnitine synthesis was required for histone acetylation and was non-redundant with other nucleo-cytosolic acetyl-CoA-generating pathways. The increase in histone acetylation by means of the αKG–carnitine axis promoted HR-mediated DNA repair through site-specific histone acetylation. Finally, we observed a positive correlation between TMLHE and histone acetylation in patient samples and found that high TMLHE or acetylcarnitine correlates with worse progression-free survival in patients treated with DNA-damaging agents. This study demonstrates for the first time, to our knowledge, that αKG affects site-specific histone acetylation and provides a mechanism of HR proficiency through carnitine synthesis. Moreover, these data provide a metabolic avenue for inducing HR deficiency and promoting sensitivity to DNA-damaging agents.
Similar content being viewed by others
KDM3A catalyses the oxidation of acetyl-lysine to hydroxyacetyl-lysine on histone H3K9

Lactylation as a metabolic-epigenetic switch in cancer: dual roles in cell death resistance and therapeutic vulnerability

Main
The ability of cells to repair DNA damage accurately and efficiently, especially DNA double-strand breaks (DSBs), is crucial for genome stability3. In cancer, DNA-repair pathways are often dysregulated4,5. HR is deficient in many tumours, leading to increased sensitivity to DNA-damaging agents that are used widely in clinical settings. Notably, HR-proficient tumours are often harder to treat, owing to their intrinsic resistance to DNA-damaging agents1. For instance, tumours with high endogenous CCNE1 expression, which encodes the oncogene cyclin E1, are HR proficient and inherently resistant to DNA-damaging agents6. The mechanisms underlying HR proficiency in general, and in the context of CCNE1 and other oncogenes, remain unclear.
Histone tail post-translational modifications (PTMs) have a crucial role in many fundamental cellular processes, including DNA DSB repair7,8,9. Histone PTMs are metabolically sensitive10. For instance, αKG, also known as 2-oxoglutarate (2OG), is a co-substrate for αKGDDs that include DNA and histone demethylases2. αKGDDs are a superfamily of enzymes that reside at the intersection of metabolism and epigenetics, and have crucial roles in many biological processes. They catalyse hydroxylation reactions on various substrates, consuming αKG and producing succinate. Alteration of several metabolic enzymes and pathways that affect αKG abundance and αKGDD activity have been reported in cancer, such as mutations of the genes encoding isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2), changes in glutaminolysis that deplete αKG, and alterations in fumarate hydratase and succinate dehydrogenase that inhibit αKGDD activity11. Previous work has found that oncometabolites that suppress αKGDD activity affect DNA repair and sensitivity to DNA-damaging agents12,13,14,15,16,17,18. So far, most studies on αKG and αKGDDs have focused on the handful of DNA and histone demethylases. However, because there are more than 60 αKGDDs, there is a strong likelihood that changes in αKG abundance also affect DNA-repair processes through other mechanisms. Our studies identify a histone acetylation pathway promoted by αKG production that is indispensable for HR proficiency and corresponding resistance to DNA-damaging agents.
αKG drives drug resistance via TMLHE
We previously showed that wild-type IDH1 and αKG are increased in high-grade serous ovarian cancer19. On further analysis, we found that the high-grade serous ovarian cancer cell lines used have high expression of CCNE1 (encoding cyclin E1). Thus, we aimed to determine whether CCNE1 drives αKG abundance. Overexpression of CCNE1 increased αKG abundance in two isogenic cell lines (Fig. 1a and Extended Data Fig. 1a). αKG is derived from multiple metabolic pathways (Fig. 1b). Depletion of αKG using either an IDH1 inhibitor (Fig. 1a) or glutamine starvation (Extended Data Fig. 1b) sensitized CCNE1-driven cells to IC10–20 doses of the DNA-damaging agents olaparib (a poly(ADP-ribose) polymerase (PARP) inhibitor), cisplatin or gamma radiation (Fig. 1c,d and Supplementary Fig. 1a,b). The IC10–20 doses were experimentally derived and chosen to observe synergy (see Supplementary Table 1 for concentrations). Inhibition of IDH1 also sensitized CCNE1-driven tumours to olaparib in vivo (Fig. 1e). Inhibition of IDH1 did not sensitize CCNE1-driven cells to the microtubule-stabilizing agent paclitaxel (Supplementary Fig. 1c). Wild-type IDH1 overexpression fully rescued the phenotype, demonstrating that the observations are not due to off-target effects (Extended Data Fig. 1c,d). Although the IDH1 inhibitor decreased both αKG and citrate abundance (Fig. 1a and Extended Data Fig. 1e), supplementing with αKG, but not citrate, rescued the observed sensitivity to DNA-damaging agents in CCNE1-driven cells (Fig. 1f and Supplementary Fig. 1d). Similarly, glutamine starvation was rescued by αKG supplementation (Fig. 1g and Supplementary Fig. 1e). To determine whether other oncogenes phenocopied CCNE1, we overexpressed MYC, which increases αKG abundance20,21 (Extended Data Fig. 1f,g). Indeed, IDH1 inhibition or glutamine starvation in MYC-overexpressing cells decreased αKG abundance (Extended Data Fig. 1h,i) and sensitized MYC-driven cells to DNA-damaging agents, which was rescued by supplementation of αKG (Extended Data Fig. 1j). Furthermore, supplementation of αKG to Kuramochi cells, which are sensitive to DNA-damaging agents, also partly reversed sensitivity to both olaparib and cisplatin (Extended Data Fig. 1k,l). Although control FT282 cells were not sensitized to cisplatin by the suppression of αKG, we did find that supplementation of αKG modestly increased cisplatin IC50 (Extended Data Fig. 1m), indicating that αKG-induced therapeutic resistance is not simply due to a CCNE1- or MYC-driven replication stress response. Together, these results demonstrate that αKG drives resistance to DNA-damaging agents.
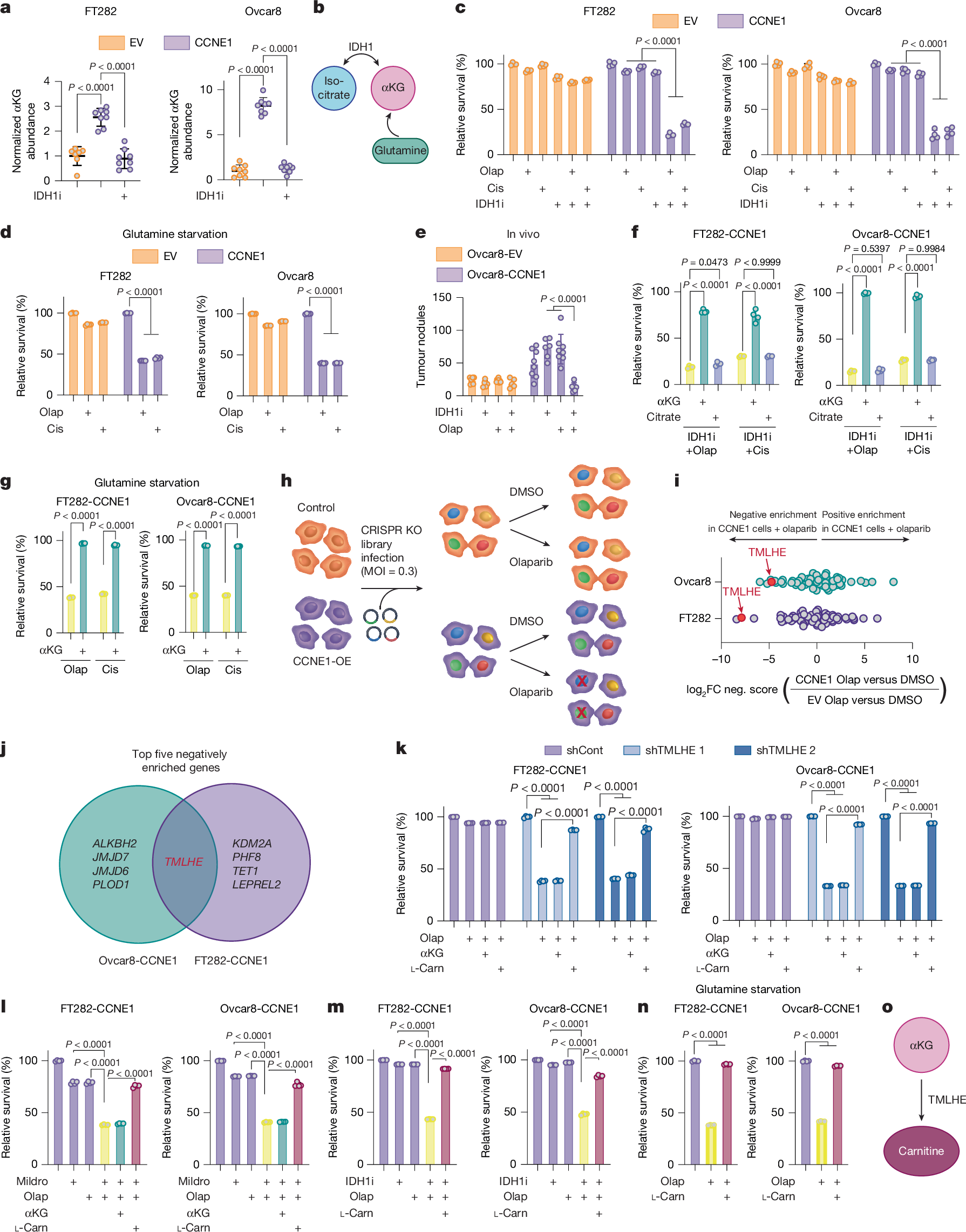
a, αKG abundance in cells expressing empty vector (EV) or CCNE1 treated with the IDH1 inhibitor (IDH1i) (EV, n = 7; CCNE1, n = 8; CCNE1 + IDH1i, n = 8; technical replicates are representative of three independent experiments in each cell-line pair). FT282 and Ovcar8 are cell types. b, Schematic of the αKG synthesis relevant to this study. c,d, Cells were treated with the IDH1 inhibitor (c) or cultured in glutamine-free media (d) and treated with olaparib (Olap) or cisplatin (Cis) either alone or in combination. e, Cells were injected intraperitoneally into mice. Mice were treated with vehicle (EV, n = 7; CCNE1, n = 8), IDH1i (EV, n = 5; CCNE1, n = 6) and olaparib alone (EV, n = 8; CCNE1, n = 8) or in combination (EV, n = 5; CCNE1, n = 6). Tumour burden was calculated by counting peritoneal tumour nodules at the end point. f,g, Cells were treated with the IDH1 inhibitor (f) or cultured in glutamine-free media (g) and treated with olaparib or cisplatin alone and in combination with αKG or citrate. h,i, Schematic (h) and analysis (i) of CRISPR knockout (KO) screen. MOI, multiplicity of infection; OE, overexpession. j, Venn diagram of the top five negatively enriched genes in the CRISPR KO screen in both CCNE1-high cell lines. FC, fold change. k,l, Cells were transduced with shGFP (shCont) or shRNAs targeting TMLHE (shTMLHE) (k) or treated with mildronate (Mildro; l) and treated with olaparib alone and supplemented with αKG or l-carnitine (l-Carn). m,n, Cells were treated with the IDH1 inhibitor (m) or cultured in glutamine-free media (n) and treated with olaparib alone and in combination with or without l-carnitine. o, Schematic of αKG upstream of TMLHE and carnitine. In c,d,f,g and k–n, percentage relative survival was normalized to vehicle controls. In c,d,f,g and k–n, n = 4 technical replicates representative of three independent experiments for each cell line. Graphs represent mean ± s.d. One-way analysis of variance (ANOVA) was used followed by Sidak’s multiple comparisons test.
αKG is required as a co-substrate for αKGDDs in mammalian cells, and αKGDDs are inhibited by succinate2. Succinate supplementation was a phenocopy of decreased αKG in its ability to sensitize CCNE1-driven cells to DNA-damaging agents and was rescued by competing αKG supplementation (Extended Data Fig. 2a), indicating that this effect is due to decreased activity of αKGDDs. To ascertain which αKGDD is driving the observed phenotype, we constructed a CRISPR knockout library of 64 αKGDDs and performed a dropout screen in the presence of olaparib (Fig. 1h). TMLHE, which encodes the first and rate-limiting enzyme in de novo carnitine synthesis, was the only one of the top five genes to drop out in both CCNE1-driven cell lines (Fig. 1i,j, Supplementary Fig. 2a and Supplementary Table 2). Using TMLHE short hairpin RNA (shRNA) (Extended Data Fig. 2b,c) and the carnitine synthesis inhibitor mildronate, we validated this observation in both CCNE1-driven cell lines using both olaparib and cisplatin (Fig. 1k,l and Extended Data Fig. 2d,e), whereas empty-vector control cells were not sensitized to DNA-damaging agents by TMLHE knockdown or mildronate (Extended Data Fig. 2d,e and controls in Supplementary Fig. 2b–d). Supplementing TMLHE-knockdown cells or mildronate-treated cells with l-carnitine, but not αKG, reversed sensitivity to DNA-damaging agents, confirming that αKG was upstream of TMLHE-catalysed carnitine synthesis (Fig. 1k,l and Extended Data Fig. 2d,e). Similarly, supplementation of l-carnitine in combination with αKG-depleting conditions or succinate reversed sensitivity to olaparib or cisplatin (Fig. 1m,n, Extended Data Figs. 1j and 2f–h). Finally, supplementation of l-carnitine was sufficient to partly rescue the sensitivity to DNA-damaging agents in Kuramochi cells (Extended Data Fig. 2i) and modestly increased cisplatin IC50 in FT282 parental cells (Extended Data Fig. 1m). Together, these data place carnitine downstream of αKG and demonstrate the necessity of the αKGDD TMLHE and the de novo carnitine-synthesis pathway for resistance to DNA-damaging agents in HR-proficient models (Fig. 1o).
αKG increases carnitine synthesis
Next, we aimed to determine the contribution of αKG to carnitine abundance and synthesis (schematic in Fig. 2a). We first measured metabolite abundance in cells that were depleted of αKG by glutamine starvation or treatment with the IDH1 inhibitor (Fig. 1a and Extended Data Fig. 1b). Depletion of αKG corresponded to decreased abundance of l-carnitine, as well as acetylcarnitine and propionylcarnitine, which was rescued by supplementation with αKG (Fig. 2b–e and Extended Data Fig. 3a,b). Total carnitine abundance was significantly affected by αKG (Extended Data Fig. 3c,d). Furthermore, inhibition of IDH1 in CCNE1-driven tumours in vivo decreased the abundance of αKG, l-carnitine, and acetylcarnitine (Fig. 2f). We also observed a positive correlation between αKG and l-carnitine/acetylcarnitine using cell-line data from DepMap (Extended Data Fig. 3e) and an increase in l-carnitine abundance in MYC-overexpressing cells, which was decreased by both inhibition of IDH1 and glutamine starvation (Extended Data Fig. 3f). TMLHE converts trimethyllysine (TML) to hydroxytrimethyllysine (HTML) and requires αKG (Fig. 2a). Thus, we used the HTML:TML ratio as a proxy for TMLHE activity. The HTML:TML ratio was decreased by IDH1 inhibition and rescued by αKG (Extended Data Fig. 3g). Towards understanding the contribution of αKG to carnitine synthesis, we performed tracing experiments in FT282-CCNE1 cells. Tracing into carnitine through the de novo pathway is technically challenging owing to a lack of commercially available stable carbon isotope-labelled analogue to trace carbons derived from trimethyllysine into carnitine. Therefore, we used a deuterated trimethyllysine (d9-TML) (Fig. 2g). We observed a decrease in d9-HTML:d9-TML in FT282-CCNE1 cells treated with the IDH1 inhibitor, and this was rescued by αKG (Fig. 2h). Together, our data demonstrate that cellular carnitine abundance and HTML synthesis through TMLHE and the de novo carnitine-synthesis pathway are regulated in an αKG-dependent manner.
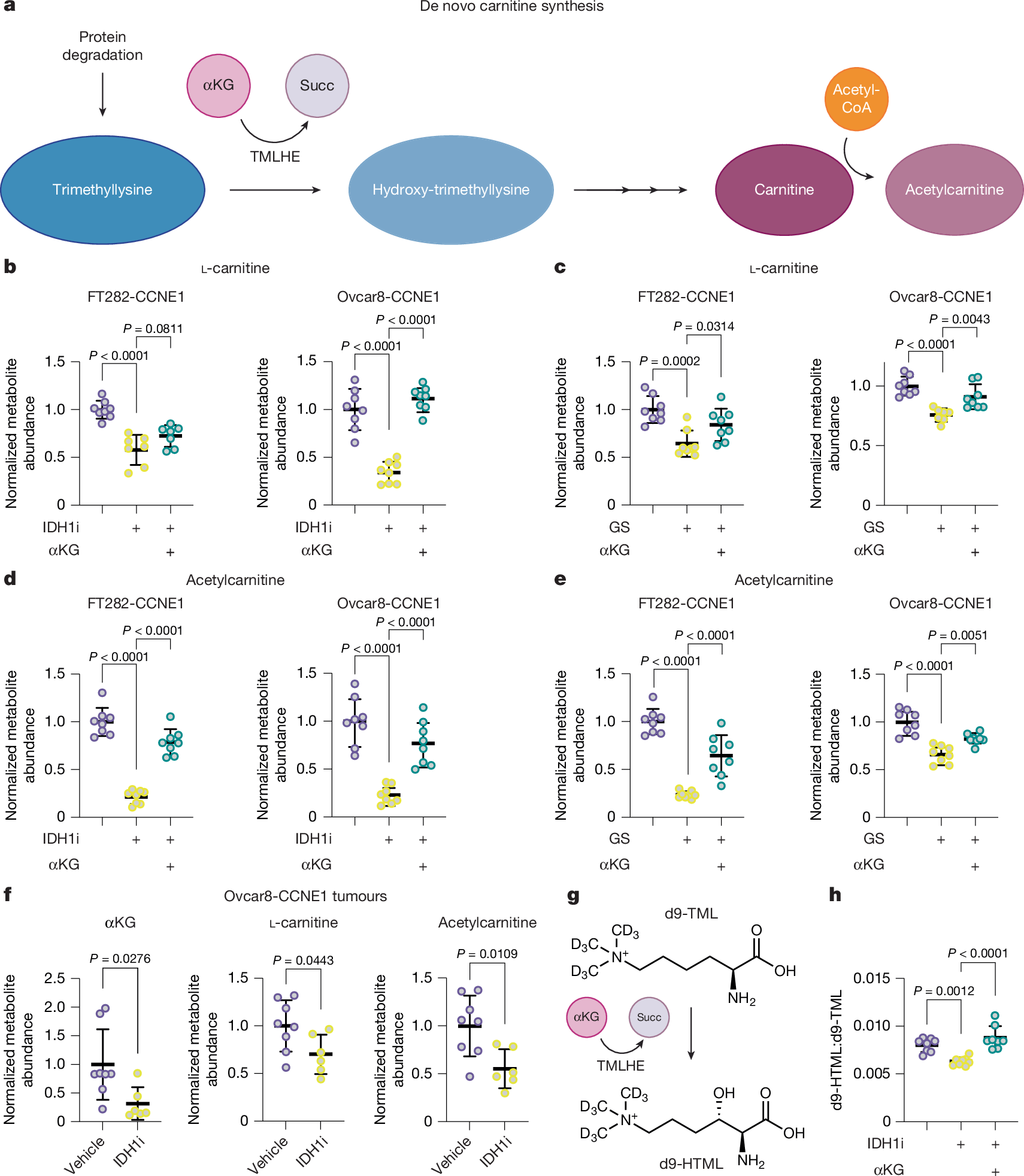
a, Schematic of the de novo carnitine-synthesis pathway. Succ, succinate. b, Carnitine abundance was assessed in cells treated with IDH1i and supplemented with αKG (FT282-CCNE1: control, n = 8; IDH1i, n = 7; IDH1i + αKG, n = 7; Ovcar8-CCNE1, n = 8; technical replicates are representative of three independent experiments for each cell-line pair). c, l-Carnitine abundance was assessed in cells cultured in normal media or glutamine starved (GS) supplemented with αKG (FT282-CCNE1: n = 8; Ovcar8-CCNE1: control, n = 8; IDH1i, n = 8; IDH1i + αKG, n = 7; technical replicates are representative of three independent experiments for each cell-line pair). d, Same as b, but acetylcarnitine was assessed (n = 8 technical replicates are representative of three independent experiments for each cell line. e, Same as c, but acetylcarnitine was assessed (n = 8 technical replicates representative of three independent experiments for each cell line). f, Ovcar8-CCNE1 cells were injected intraperitoneally into mice. Mice were treated with vehicle (n = 8 per group) or the IDH1 inhibitor (n = 6 per group). At the end point, omental tumours were collected for analysis of the indicated metabolites. g, Schematic of d9-TML tracing into d9-HTML. h, The d9-HTML:d9-TML ratio was assessed in FT282-CCNE1 cells treated with the IDH1i alone or supplemented with αKG (n = 8 technical replicates representative of two independent experiments). Graphs represent mean ± s.d. In b–e and h, one-way ANOVA was followed by Sidak’s multiple comparisons test. In f, two-tailed Student’s t-test.
αKG drives histone acetylation
Carnitine is needed to transport short and long acyl groups across the inner mitochondrial membrane, and acylcarnitines are known to be important for both energy production and redox balance22,23. Surprisingly, l-carnitine and acetylcarnitine, but not propionylcarnitine or butyrylcarnitine, rescued the sensitivity to DNA-damaging agents in combination with IDH1 inhibition, glutamine starvation, TMLHE knockdown, carnitine synthesis inhibition or succinate supplementation (Supplementary Fig. 3). The antioxidant n-acetyl-l-cysteine (NAC) also did not rescue the observed effects (Supplementary Fig. 4). Because experiments using propionylcarnitine/butyrylcarnitine or NAC did not phenocopy the l-carnitine supplementation, these data indicate that cellular energetics and/or reactive oxygen species do not contribute to the observed effects.
Acetylcarnitine is a precursor for nuclear acetyl-CoA that supports histone acetylation24,25. We found that knockdown or inhibition of IDH1 decreased global histone acetylation more than histone methylation (Fig. 3a and Supplementary Table 3). We also observed decreased pan-acetylation of histones H3 and H4 in Ovcar8-CCNE1 tumours in mice treated with the IDH1 inhibitor (Fig. 3b and Extended Data Fig. 4a). These data point to a previously unrecognized effect of αKG-mediated epigenetic rewiring through histone acetylation. Elevated histone acetylation was confirmed in an additional cell line and shown to decrease through the depletion of αKG or the suppression of αKGDDs usi