Genome modelling and design across all domains of life with Evo 2
TL;DR
Evo 2 is a biological foundation model trained on 9 trillion DNA base pairs across all domains of life, enabling zero-shot prediction of genetic variant effects and generative genome design. It uses a 1 million token context window and open-source resources to advance computational biology.
Key Takeaways
- •Evo 2 learns from diverse genomic data to predict functional impacts of mutations without task-specific fine-tuning.
- •The model generates realistic mitochondrial, prokaryotic, and eukaryotic sequences with high coherence.
- •It achieves accurate zero-shot variant effect prediction for both coding and noncoding human DNA.
- •Evo 2's embeddings support lightweight classifiers for tasks like exon identification and gene essentiality.
- •The model and its training dataset are fully open-source to accelerate biological research and design.
Tags
Abstract
All of life encodes information with DNA. Although tools for genome sequencing, synthesis and editing have transformed biological research, we still lack sufficient understanding of the immense complexity encoded by genomes to predict the effects of many classes of genomic changes or to intelligently compose new biological systems. Artificial intelligence models that learn information from genomic sequences across diverse organisms have increasingly advanced prediction and design capabilities1,2. Here we introduce Evo 2, a biological foundation model trained on 9 trillion DNA base pairs from a highly curated genomic atlas spanning all domains of life to have a 1 million token context window with single-nucleotide resolution. Evo 2 learns to accurately predict the functional impacts of genetic variation—from noncoding pathogenic mutations to clinically significant BRCA1 variants—without task-specific fine-tuning. Mechanistic interpretability analyses reveal that Evo 2 learns representations associated with biological features, including exon–intron boundaries, transcription factor binding sites, protein structural elements and prophage genomic regions. The generative abilities of Evo 2 produce mitochondrial, prokaryotic and eukaryotic sequences at genome scale with greater naturalness and coherence than previous methods. Evo 2 also generates experimentally validated chromatin accessibility patterns when guided by predictive models3,4 and inference-time search. We have made Evo 2 fully open, including model parameters, training code5, inference code and the OpenGenome2 dataset, to accelerate the exploration and design of biological complexity.
Main
Biological research spans scales from molecules to systems to organisms, seeking to understand and design functional components across all domains of life. Creating a machine to design functions across the diversity of life would require it to learn a deep, generalist representation of biological complexity. Although this complexity surpasses straightforward human intuition, advances in artificial intelligence offer a universal framework that leverages data and compute at scale to uncover higher-order patterns6,7. We reasoned that training a model with these capabilities would require data spanning the full spectrum of biological diversity to discover emergent properties similar to those found in other fields8.
We previously demonstrated that machine learning models trained on prokaryotic genomic sequences can model the function of DNA, RNA and proteins, as well as their interactions that create complex molecular machines1,2. Here we present Evo 2, a biological foundation model trained on a representative snapshot of genomes spanning all domains of life. We extend the sequence modelling paradigm to the scale and complexity of eukaryotic genomes through advances in data curation, model architecture, large-scale pre-training, advanced interpretability methods and inference-time prediction and generation approaches.
Emphasizing generalist capabilities over task-specific optimization, Evo 2 represents an important milestone in biological sequence modelling, laying a broad foundation for prediction and design tasks that are relevant to all modalities of the central dogma, that span molecular to genome scale and that generalize across all domains of life.
Evo 2 architecture, training, and data
Evo 2 was trained on prokaryotic and eukaryotic genetic sequences, with potential downstream utility for predictive and generative tasks across multiple scales of complexity (Fig. 1a). We trained two versions of Evo 2: a smaller version with 7 billion parameters trained on 2.4 trillion tokens (Evo 2 7B), and a larger version with 40 billion parameters trained on 9.3 trillion tokens (Evo 2 40B). This new training dataset, which we call OpenGenome2, was compiled from curated, non-redundant nucleotide sequence data with a total of more than 8.8 trillion nucleotides from bacteria, archaea, eukarya and bacteriophage (Fig. 1b and Extended Data Fig. 1a).
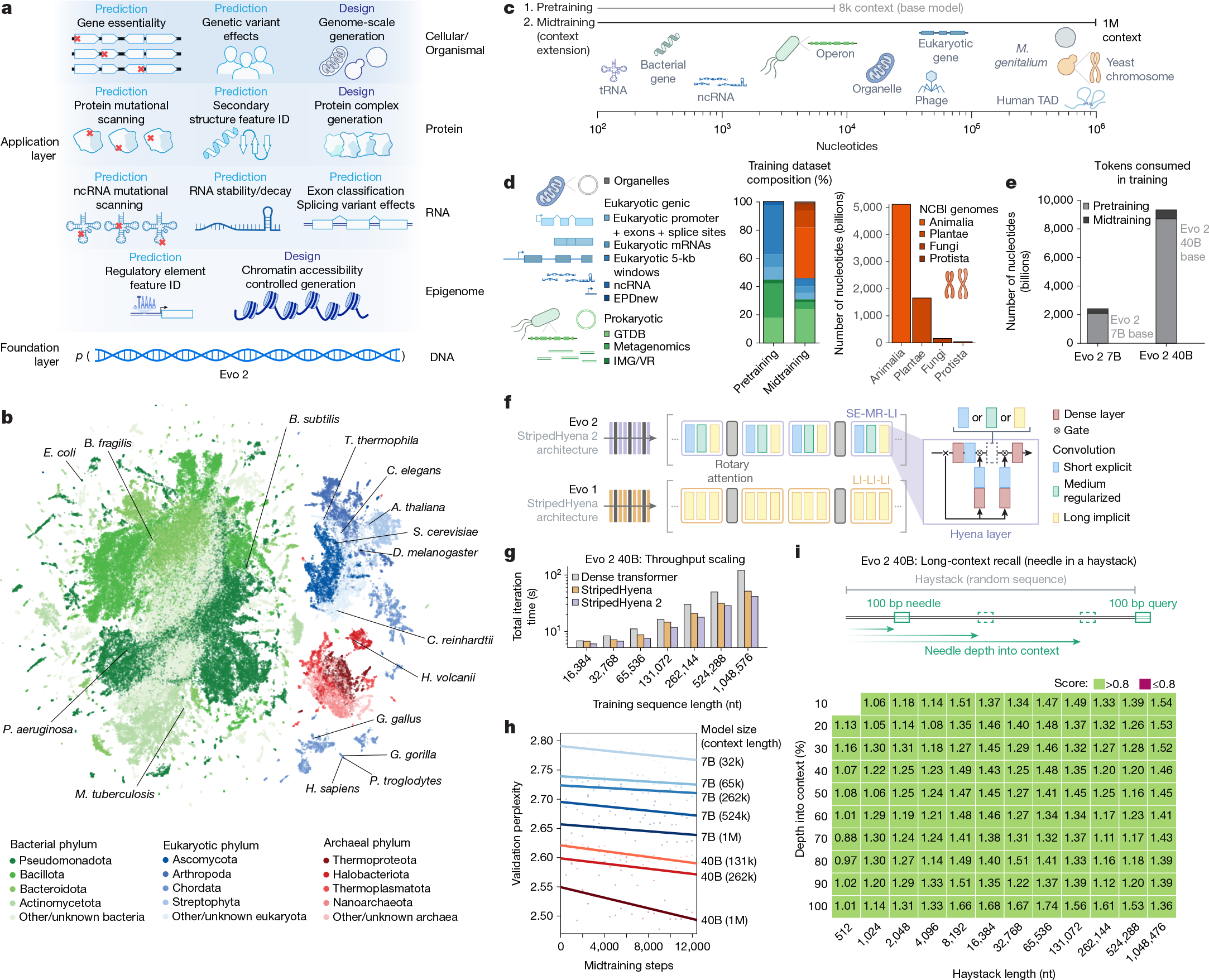
a, Evo 2 models DNA sequence and enables applications across the central dogma, scaling from molecules to genomes and spanning all domains of life. b, Evo 2 was trained on data encompassing trillions of nucleotide sequences from all domains of life. Each point in the UMAP (uniform manifold approximation and projection) graph represents a single genome in the training dataset that is embedded on the basis of the genome’s k-mer frequencies. Arabidopsis thaliana, Bacillus subtilis, Bacteroides fragilis, Caenorhabditis elegans, Chlamydomonas reinhardtii, D. melanogaster, E. coli, Gallus gallus, Gorilla gorilla, Haloferax volcanii, Homo sapiens, Mycobacterium tuberculosis, Pan troglodytes, Pseudomonas aeruginosa, S. cerevisiae and Tetrahymena thermophila are highlighted. c, A two-phase training strategy was used to optimize model performance while expanding the context length up to 1 million base pairs to capture wide-ranging biological patterns. M. genitalium, Mycoplasma genitalium; TAD, topologically associating domain. d, Novel data augmentation and weighting approaches prioritize functional genetic elements during pretraining and long-sequence composition during midtraining. GTDB, Genome Taxonomy Database; IMG/VR, Integrated Microbial Genomes/Virus database. e, The number of tokens used to train Evo 2 40B and 7B, split into the shorter sequence pretraining and the long context midtraining. f, Schematic of the new multi-hybrid StripedHyena 2 architecture, showing the efficient block layout of short explicit (SE), medium regularized (MR) and long implicit (LI) hyena operators. g, Comparison of iteration time at 1,024 GPU, 40B scale between StripedHyena 2, StripedHyena 1 and Transformers, showing improved throughput. h, Validation perplexity of Evo 2 midtraining comparing the model size and context length, showing benefits with scale and increasing context length. i, A modified needle-in-a-haystack task was used to evaluate long context recall ability up to 1 million sequence length, and shows that Evo 2 performs effective recall at 1 million token context.
Both Evo 2 7B and 40B are trained in two phases to capture biological length scales from molecular to organismal (Fig. 1c–e). Our first stage of pretraining uses a context length of 8,192 tokens, with data weighting focused on genic windows to learn functional genetic elements, followed by a multi-stage midtraining phase over which we extend the context length of Evo 2 to 1 million tokens to learn the relationships between elements across long genomic distances (Fig. 1c–e and Methods). This matches best practice in natural language, in which initial pretraining at shorter context lengths improves both efficiency and overall model quality9,10,11. As in Evo 1, we excluded genomic sequences from viruses that infect eukaryotic hosts from the training data for biosafety purposes. We verified that these data exclusions led to high perplexity on genomic sequences from eukaryotic viruses (Extended Data Fig. 2a), indicating poor language modelling performance in this domain.
Evo 2 uses StripedHyena 2, a convolutional multi-hybrid architecture5 that relies on a combination of three different variants of input-dependent convolution operators12 and attention (Fig. 1f and Extended Data Fig. 1b), improving training efficiency at scale on both short and long sequences, as well as allowing each layer to model interactions at variable distances. StripedHyena 2 provides substantially higher throughput (at 40 billion parameters, up to 3× speedup at 1 million context length) than highly optimized Transformer6 baselines and previous generation hybrid models based on recurrences or long convolutions, such as StripedHyena 1 (ref. 13) (Fig. 1g). StripedHyena 2 also improves loss scaling on DNA against both Transformers and StripedHyena 1 (Extended Data Fig. 1c), thereby achieving both lower prediction error with the same amount of training data and enabling more efficient use of computational resources.
We train up to 1 million base pairs in context length through a multi-stage extension phase, which showed improvements in loss with both model scale and longer context (Fig. 1h). With a synthetic long-context evaluation called ‘needle-in-a-haystack’, we show that Evo 2 can identify and predict the value of a specific 100 base pair sequence (the needle) hidden within 1 million base pairs of random DNA (the haystack), serving as a synthetic quality check that the model can retrieve information from its full context window, as desired for long-context models (Fig. 1i and Extended Data Fig. 1d,e).
Evo 2 learns evolutionary constraint
By learning the likelihood of sequences across vast evolutionary datasets, biological sequence models capture conserved sequence patterns that often reflect functional importance. These constraints allow the models to perform zero-shot prediction without any task-specific fine-tuning or supervision1,14,15,16. Here, likelihood refers to the probability that the model assigns to a given sequence, where mutations that reduce this probability are predicted to be deleterious. Given that Evo 2 learns a likelihood landscape across all three modalities of the central dogma (DNA, RNA and protein) and all three domains of life, we sought to assess whether Evo 2 could perform mutational effect prediction across these modalities and organisms (Fig. 2a).
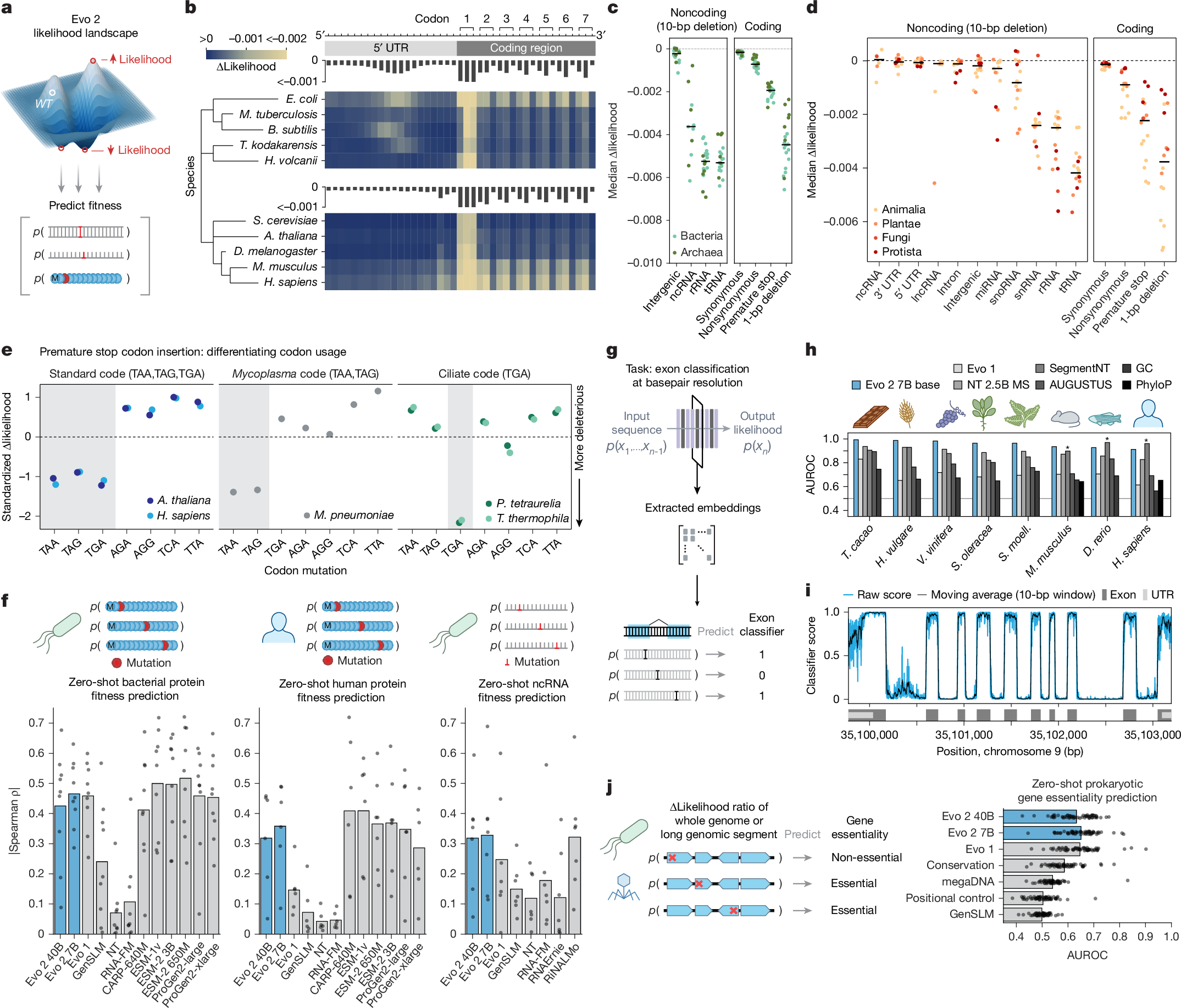
a, Evo 2-predicted zero-shot likelihoods can be used to predict the effects of DNA, RNA or protein mutations on molecular function or organismal fitness. WT, wild type. b, Effects on Evo 2 prediction of sequence likelihood caused by mutations along gene start sites for various model species across the domains of life. See Extended Data Fig. 3a,b for additional analyses. T. kodakarensis, Thermococcus kodakarensis. c,d, For different prokaryotic (c) and eukaryotic (d) sequences, the likelihood of different types of mutations in different genomic elements were scored using Evo 2 7B. Scatter represents the median change in likelihood from wild type to mutant sequence per species, coloured by domain (c) or kingdom (d). Horizontal line indicates the median of the scatter distribution. lncRNA, long noncoding RNA; snRNA, small nuclear RNA. e, Mutational likelihoods were used to assess the ability of Evo 2 to differentiate between genomic sequences of model organisms on the basis of their usage of different stop codons. Shown are the standardized median of delta likelihood values across 5 species, where medians were calculated across approximately 4,100 randomly selected mutation loci. M. pneumoniae, Mycoplasma pneumoniae; P. tetraurelia, Paramecium tetraurelia. f, DMS assays were used to assess the Spearman correlation of zero-shot likelihoods from models with experimental assays. Notably, Evo 1 and GenSLM were exclusively trained on prokaryotic datasets. g, Schematic of our single-nucleotide resolution exon classifier based on embeddings from Evo 2. h, Single-nucleotide exon classifiers were trained on embeddings from Evo 2, Nucleotide Transformer (NT) and Evo 1, and were evaluated on the basis of their AUROC across eight held-out species. Performance was compared to SegmentNT-30 kb multispecies (asterisks indicate species in SegmentNT training data), ab initio AUGUSTUS, and to baseline nucleotide content and conservation metrics. D. rerio, Danio rerio; H. vulgare, Hordeum vulgare; S. moell., Selaginella moellendorffii; S. oleracea, Spinacia oleracea; T. cacao, Theobroma cacao; V. vinifera, Vitis vinifera. i, Genome browser track showing predictions from the Evo 2 embedding-based exon classifier scanned across the human STOML2 locus, where the vertical axis is the predicted classifier score and the horizontal axis is genome position. j, Evo 2 predicts genes as essential or nonessential, as determined by experimental gene essentiality assays across bacterial, archaeal and phage species (shown as overlaid scatter) using mutational likelihood of premature stop codon insertions (as a genetic perturbation).
To assess whether Evo 2 captures core biological principles, we first evaluated how single nucleotide variants (SNVs) affect Evo 2 likelihoods in the genomic sequences around the start codons of protein-coding genes. We introduced these mutations at each position in the wild-type sequence and calculated the resulting changes in Evo 2 predicted likelihoods across thousands of such loci (Fig. 2b and Extended Data Fig. 3a). We observed strong changes in the likelihood for mutations within the start codons in both prokaryotes and eukaryotes. This was followed by a three-base periodicity pattern reflecting the triplet codons, with changes at the wobble positions showing lower impact on likelihood. For both prokaryotic and eukaryotic genomes, we observed a pattern upstream of the coding DNA sequence (CDS) that was consistent with the locations of known consensus sequences associated with translation initiation, namely, the Shine–Dalgarno sequence17 for prokaryotes and the Kozak sequence18 for eukaryotes. We also observed similar patterns for SNVs around stop codons (Extended Data Fig. 3b).
Next we measured the effect of mutations across a variety of both noncoding and coding sequences (Fig. 2c,d). Across 20 prokaryotic species and 16 eukaryotic species, we observed changes in model likelihoods consistent with known biological constraints. Non-synonymous mutations, premature stop codons and frameshift mutations caused much larger changes in likelihood than synonymous mutations. In noncoding regions, deletions in transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) had much larger effects than deletions in intergenic and other noncoding loci, reflecting the known essential roles of these RNAs. The 40B model exhibited higher sensitivity to deletions in microRNA (miRNA) and small nucleolar RNA (snoRNA) sequences compared with the 7B model. Evo 2 also predicted that less efficiently translated codons had lower likelihoods than more efficient codons (Extended Data Fig. 3c–e).
Recognizing that our training data contained genomes with distinct genetic codes, we tested how different premature stop codons impacted species that differ in their stop codon usage (Fig. 2e). We found that the model learned the difference between the standard code (stop codons TAA, TAG and TGA), the mycoplasma code (Code 4, stop codons TAA and TAG) and the ciliate code (Code 6, stop codon TGA). When ciliate genomes were artificially recoded to the standard genetic code, Evo 2 predicted mutations from the standard stop codons as deleterious, demonstrating that the model relies on sequence context to determine the appropriate genetic code (Extended Data Fig. 3f).
Although Evo 2 likelihoods reflect the expected importance of different genetic alterations, a key question is whether these likelihoods also correlate with functional effects, which can be empirically measured via deep mutational scanning (DMS) of proteins and noncoding RNAs (ncRNAs). Although state-of-the-art methods for this task tend to leverage both sequence alignments and structural conditioning, general-purpose single-sequence protein language models also learn likelihood distributions that correlate with fitness15. Evo 2 sequence likelihoods correlate with diverse definitions of fitness across nine prokaryotic protein datasets; six eukaryotic protein datasets; and seven datasets of rRNAs, tRNAs and ribozymes (Fig. 2f). Evo 2 is competitive with widely used ProGen language models for protein DMS and with RNA language models for ncRNA DMS, although it underperforms state-of-the-art models on protein DMS. Consistent with observed trends for protein language models, the performance of Evo 2 on these fitness prediction benchmarks begins to saturate and can decrease at the largest model scales19,20,21. We also tested the ability of Evo 2 to predict mutation effects in protein sequences from viruses that infect human hosts. We found no correlation between Evo 2 likelihood and viral protein fitness (Extended Data Fig. 2b), consistent with our data exclusions having the intended effect of weakening both language modelling and downstream performance (Extended Data Fig. 2a). Evo 2 likelihoods also have modest zero-shot association with human mRNA decay rates (Extended Data Fig. 3g and Supplementary Information B.2).
Since Evo 2 learns from eukaryotic genomes, which can be challenging to annotate, we assessed whether its embeddings capture exon–intron architecture. We trained lightweight models on Evo 2 7B base embeddings to develop single-nucleotide resolution classifiers of exon labels (Fig. 2g and Methods). On eight diverse species held out from classifier training, our best classifier achieved areas under the receiver operating characteristic curve (AUROCs) ranging from 0.91 to 0.99 (Fig. 2h,i), outperforming models trained on embeddings from other genomic language models, Nucleotide Transformer22 and Evo 1 (ref. 1), and classification by conservation metrics (local GC content and PhyloP). As a practical baseline, we show that our classifier outperforms ab initio AUGUSTUS23 across all species tested. Evo 2 also outperforms SegmentNT24 on all species outside the SegmentNT training set and on one of the three species in its training set. These results suggest that combining Evo 2 sequence embeddings with supervised approaches can aid the functional annotation of genetic components across diverse species, including non-model organisms.
Beyond molecular or gene-level prediction tasks, we previously showed that high likelihood under Evo 1 is associated with whole organism replication fitness in prokaryotes and phage as quantified by gene essentiality experiments1. Using zero-shot likelihoods to score the effects of premature stop codon insertions into bacterial, archaeal and phage genomes, we found that Evo 2 models performed similarly to Evo 1 and better than other zero-shot methods in predicting gene essentiality across diverse species (Fig. 2j and Extended Data Fig. 3h). On zero-shot prediction of human gene essentiality (Methods), Evo 2 40B (AUROC = 0.66, area under the precision-recall curve (AUPRC) = 0.15) outperformed other genomic language models (AUROC range 0.50–0.59, AUPRC range 0.09–0.12) and performs within the range of four PhyloP conservation scores (AUROC range 0.65–0.71, AUPRC range 0.13–0.21) (Extended Data Fig. 3i), although the overall predictive performance remains modest.
Together, these results demonstrate that Evo 2 captures information across biological modalities and domains of life. Notably, the 7B and 40B models expand predictive capabilities without compromising the prokaryotic insights captured by Evo 1. The utility of both zero-shot likelihoods and simple classifiers trained on Evo 2 embeddings for a variety of predictive tasks across prokaryotic and eukaryotic genomes indicates that Evo 2 provides a strong foundation model for downstream applications in computational biology.
Human variant effect prediction
Variant effect prediction represents a critical challenge in genomics, with direct implications for clinical diagnosis and therapeutic development. Genomic language models have previously struggled in eukaryotic variant effect prediction, lagging considerably behind species-specific models that use multiple sequence alignments16,22,25. Evo 2 can perform accurate zero-shot variant effect prediction for both coding and noncoding DNA by considering the changes in the model’s likelihoods after introducing mutations involving single or multiple nucleotides (Fig. 3a).
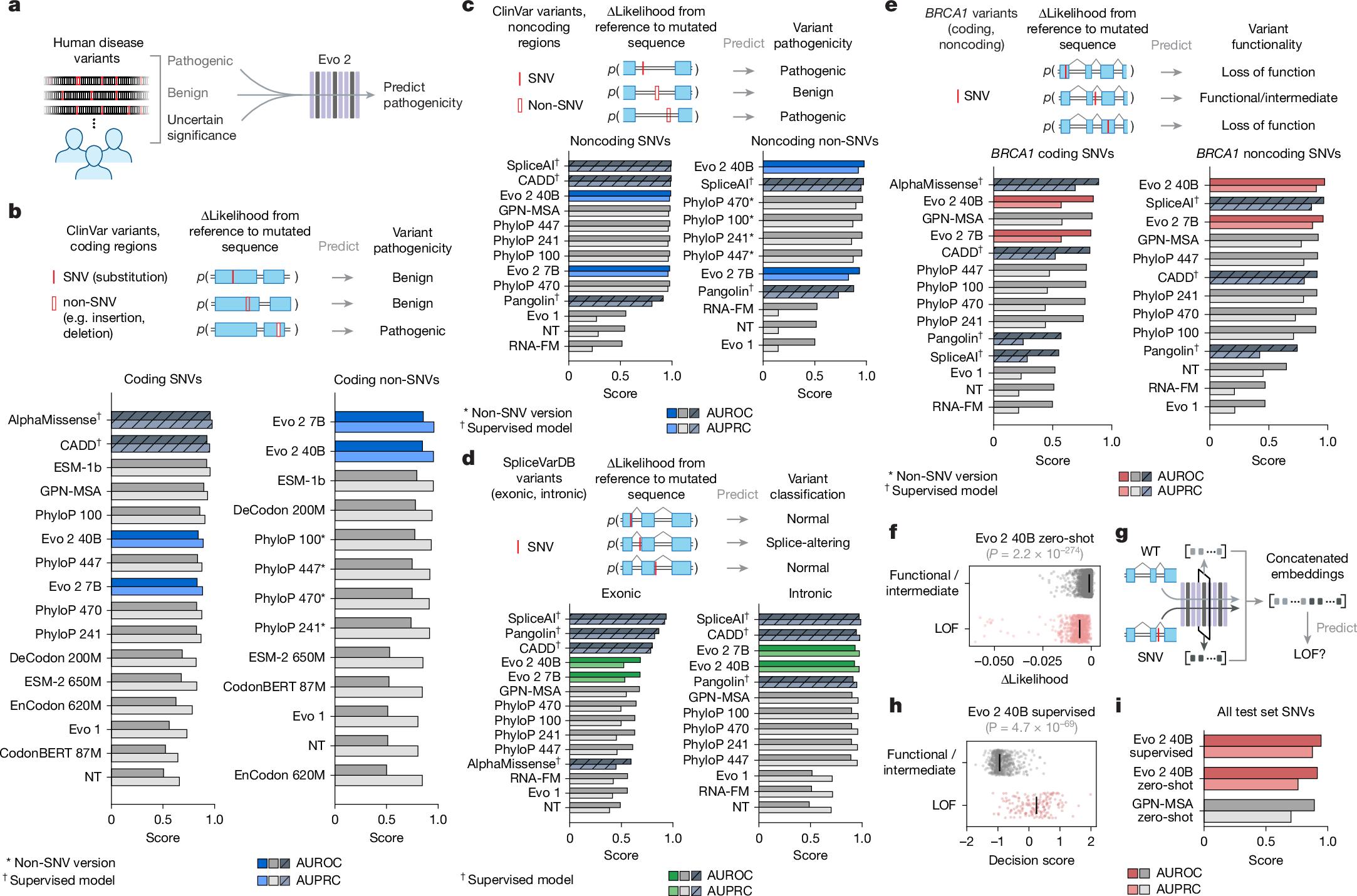
a, Overview of zero-shot variant effect prediction using Evo 2. Evo 2 was used to assign likelihood scores to human genetic variants, distinguishing pathogenic and benign variants in both coding and noncoding regions. b,c, Zero-shot evaluation of variant pathogenicity within the coding (b; n = 14,319 SNVs, n = 1,236 non-SNVs) and noncoding (c; n = 34,761 SNVs, n = 3,894 non-SNVs) regions. Shown are the AUROCs and AUPRCs for classifying pathogenic and benign variants from ClinVar, across models. For non-SNV evaluations, a modified version of PhyloP was used (Methods). d, Zero-shot evaluation on splice-altering variants in SpliceVarDB, split by exonic (n = 1,181) and intronic (n = 3,769) scoring. e, Evo 2 and other models were used to evaluate BRCA1 variant effect predictions against BRCA1 saturation mutagenesis data, comparing classification of loss-of-function versus functional and intermediate variants in both coding (n = 2,077 SNVs) and noncoding (n = 1,125 SNVs) regions. f, Evo 2 zero-shot likelihood scores plotted for loss-of-function (LOF) versus functional/intermediate variants (n = 3,893), demonstrating the ability of Evo 2 to separate these classes. P value calculated by two-sided Wilcoxon rank sum test. g, Evo 2 embeddings were extracted and concatenated to train a supervised classifier for BRCA1 variant effect prediction. h, Predictions of the supervised classifier on functional/intermediate variants compared with true loss-of-function variants on the test set (n = 789), with decision scores on the horizontal axis. P value calculated by two-sided Wilcoxon rank sum test. i, Comparison of a supervised classifier trained on Evo 2 embeddings on the BRCA1 test set against zero-shot baselines, highlighting the value of using Evo 2 embeddings to build lightweight supervised models.
We used annotations of human clinical and experimentally determined variants to evaluate the ability of Evo 2 to predict biologically important sequence variation. We also contextualize the performance of Evo 2 against a wide range of models, including statistical measures of conservation (for example, PhyloP); unsupervised language models of proteins, RNA and DNA (for example, ESM-1b); supervised splicing prediction models (for example, Pangolin and SpliceAI); and human variant effect prediction models (for example, AlphaMissense, GPN-MSA and CADD).
Using the ClinVar database, we compared the ability of Evo 2 against other methods for predicting the pathogenic effects of human genetic variants across diverse variant classes (Supplementary Data 1). For coding region SNVs, the 40B and 7B models performed competitively, ahead of zero-shot methods, including ESM-2, but behind ESM-1b, GPN-MSA and some PhyloP variants (Fig. 3b). For non-SNV coding variants (for example, insertions and deletions), both Evo 2 models outperformed all other methods; notably, these non-SNV variants are not possible to score by leading models such as AlphaMissense and GPN-MSA (Fig. 3b). For noncoding SNVs, Evo 2 40B ranked first among unsupervised models and only trailed behind supervised mod