β-Arrestin condensates regulate G-protein-coupled receptor function
Abstract
β-Arrestins 1 and 2 are multifunctional adaptor proteins1 that regulate the signalling of G-protein-coupled receptors (GPCRs), the largest class of receptors, which impact nearly all aspects of physiology and are one of the most common drug targets2. Although β-arrestins interact with a wide array of signalling effectors at many GPCRs, it is unclear how β-arrestins promote such varied functions. Here we show that β-arrestins undergo liquid–liquid phase separation, forming condensates that regulate GPCR function. We show that condensation is specific to visual arrestins and β-arrestins, and demonstrate that β-arrestin oligomerization occurs in proximity to the GPCR to regulate GPCR functions such as internalization and signalling. Our work provides a paradigm for β-arrestin condensates as regulators of GPCR function, with liquid–liquid phase separation serving as an important promoter of signalling compartmentalization at GPCRs.
Similar content being viewed by others

Helix-bundle and C-terminal GPCR domains differentially influence GRK-specific functions and β-arrestin-mediated regulation

Multi-faceted roles of β-arrestins in G protein-coupled receptor endocytosis
β-Arrestin-independent endosomal cAMP signaling by a polypeptide hormone GPCR
Main
After agonist stimulation, GPCRs activate heterotrimeric G proteins, which promote signalling through second messengers, followed by the recruitment of GPCR kinases (GRKs) and β-arrestins. Receptor phosphorylation by GRKs promotes associations with β-arrestins, which regulate GPCR desensitization, signalling and trafficking. β-Arrestins perform these multiple tasks by sterically preventing G protein activation and by acting as scaffolding proteins for the endocytic machinery and signalling molecules (for example, Raf-1, MEK1 and ERK)3,4, interacting with hundreds of proteins5. Furthermore, β-arrestins can be catalytically activated by GPCRs, followed by translocation to the plasma membrane and clathrin-coated pits (CCPs)6. β-Arrestins interact with the receptor through finger loop insertion into the receptor core and/or through binding of their N-domain to the phosphorylated receptor C terminus7,8,9. This results in the release of the β-arrestin C-tail intrinsically disordered region (IDR) from the N domain, and promotes the active conformation of β-arrestin10 characterized by interdomain twisting between the N and C domains11,12. β-Arrestins are regulated by their cellular environment through binding to specific components; for example, inositol hexaphosphate (IP6) promotes the oligomerization of β-arrestin 1 into infinite chains8 and the oligomerization of β-arrestin 2 into trimers with a conformation that is similar to β-arrestin 1 (refs. 13,14,15,16) in complex with a GPCR8,17. β-Arrestins can homo- and hetero-oligomerize18, and ablation of IP6 binding sites results in dysregulation of β-arrestin nucleocytoplasmic shuttling, signalling and protein–protein interactions19,20. However, the biological significance of β-arrestin oligomerization and whether it promotes compartmentalization of receptor signalling is unclear.
One mechanism that contributes to signalling compartmentalization by other receptor families is the formation of biomolecular condensates21,22. Biomolecular condensates are enriched with molecules that can engage in multivalent interactions through specific oligomerization motifs and IDRs23,24. These interactions reduce molecular solubility due to entropy-driven effects and promote liquid–liquid phase separation (LLPS) to produce condensates21. These condensates partition reactants to increase their local concentration and sequester signalling components22,25,26. Condensates have been described at membrane receptors such as receptor tyrosine kinases and T cell receptors26,27, in addition to their downstream effectors, such as PKA and WNK25,28. Notably, β-arrestins 1 and 2 have a C-terminal IDR and undergo oligomerization, features that are found in macromolecules enriched in biomolecular condensates.
Here we demonstrate the formation of β-arrestin condensates to regulate GPCR activity. Upon GPCR activation, we demonstrate β-arrestin oligomerization that displays an orientation dependence. Finally, we demonstrate that mutations of the IDR and residues predicted to promote oligomerization alter the ability of β-arrestins to regulate GPCR signalling and internalization.
The presence of endogenous β-arrestin condensates
When overexpressed, β-arrestin-GFP is observed in a diffuse cytosolic pattern; after agonist stimulation, β-arrestin-GFP is recruited to the plasma membrane29. To evaluate the pattern of endogenous β-arrestin expression, we used super-resolution microscopy and immunofluorescence with antibodies targeting both β-arrestins 1 and 2. In HEK293T cells, we visualized β-arrestins 1 and 2 in puncta throughout the cytosol (Fig. 1a). We validated antibody selectivity using β-arrestin 1–2 knockout (KO) cells, which had little to no observable signal (Fig. 1b). Next we assessed whether these puncta would change upon agonist stimulation. In HEK293T cells, angiotensin II (AngII) type 1 receptor (AT1R) was transfected and stimulated with AngII for 5 min. Indeed, both β-arrestin 1 and 2 puncta were largely localized to the plasma membrane (Fig. 1c). Fluorescently labelled receptors—β2 adrenergic receptor (β2AR), vasopressin receptor 2 (V2R) and AT1R—that were stimulated with their respective ligands showed β-arrestin puncta localization with receptors, although there were still puncta present in the cytosol (Extended Data Fig. 1a). We then performed immunofluorescence of overexpressed β-arrestins 1 and 2. We transfected 500 ng of untagged β-arrestin in HEK293T and, using immunofluorescence, we observed a diffuse cytosolic pattern (Fig. 1d), analogous to the diffuse cytosolic pattern observed for overexpressed β-arrestin-GFP. Overall, these findings highlight that endogenous β-arrestin puncta are recruited to the receptor and that overexpression of β-arrestin results in a diffuse cytosolic pattern.
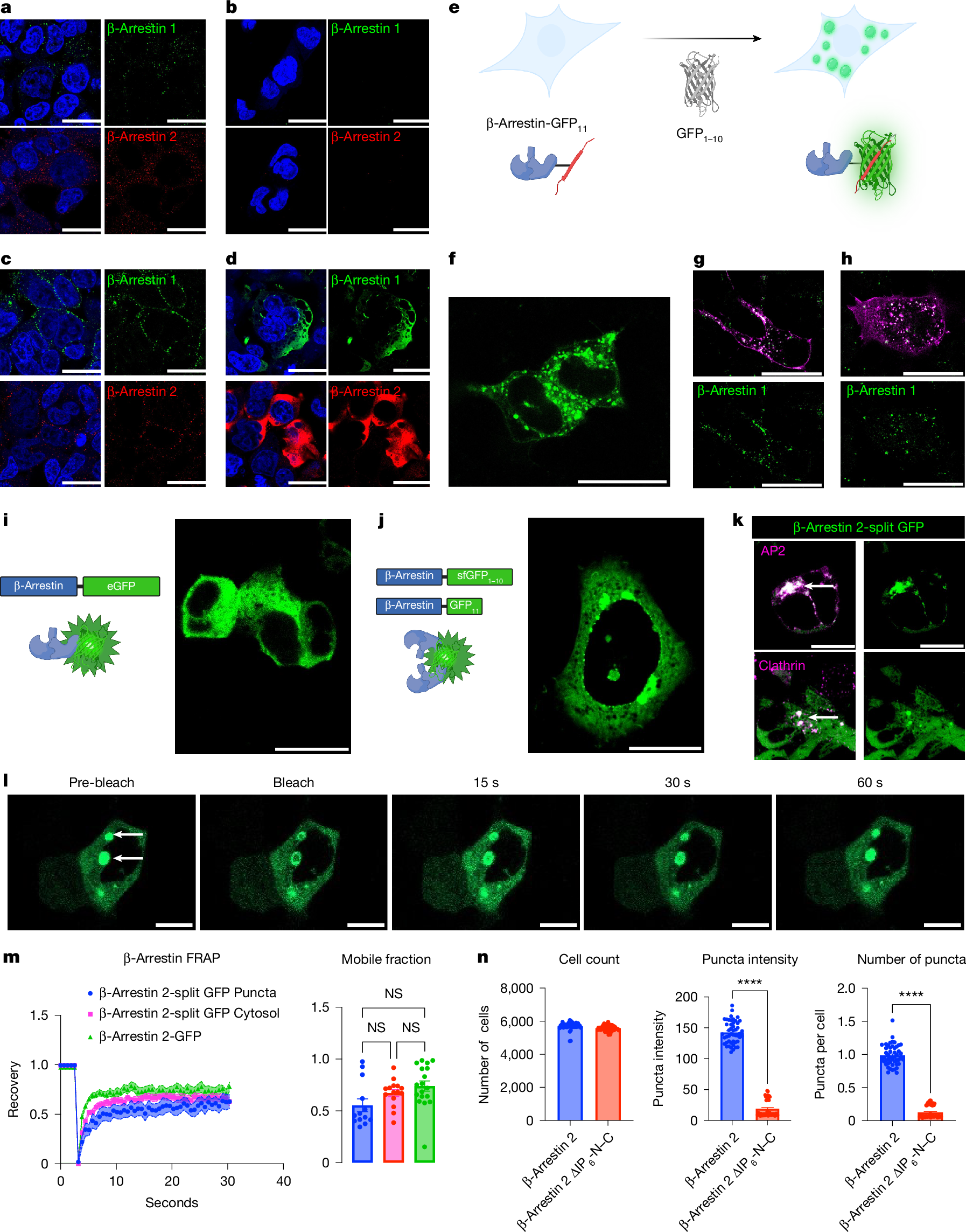
a, HEK293T cells were fixed with 4% paraformaldehyde and immunofluorescence was performed for β-arrestin 1 or 2. b, Immunofluorescence for β-arrestins 1 and 2 on β-arrestin 1–2 KO cells. c, HEK293T cells were transfected with AT1R and stimulated with AngII (1 µM) for 5 min, fixed, and then β-arrestins 1 and 2 were subjected to immunofluorescence. d, HEK293T cells were transfected with 500 ng of either β-arrestin 1 or 2, which was then subjected to immunofluorescence. e, Schematic of endogenous β-arrestin CRISPR cell line. f, Representative image of CRISPR β-arrestin 1 puncta (GFP11 + GFP1–10). g,h, CRISPR β-arrestin 1 cells were transfected with either β2AR-mKO (g) or V2R-mKO (h) and stimulated with either isoproterenol (10 µM) or AVP (1 µM), respectively. i, Diffuse cytosolic β-arrestin 2-eGFP expression in HEK293T cells transfected with a β-arrestin 2 with eGFP fused to the C terminus. j, β-arrestin 2 was fused with the 11th β-strand of GFP, or the remaining β-strands of sfGFP1–10, on the C-terminus. k, Cells were transfected with β-arrestin-2-split GFP, AP2-mKO (β-subunit) or clathrin-mKO, and were stained with DAPI. l, FRAP was performed on β-arrestin 2-split GFP puncta. m, FRAP on β-arrestin 2-split GFP puncta was compared with cytosolic split GFP and β-arrestin 2-eGFP. Curves show normalized average fluorescent intensity with s.e.m. Mobile fractions are also shown. n, Quantification of cell count, as well as the size and number of puncta with β-arrestin 2-split GFP and IP6 mutants. P ≥ 0.05, *P = 0.01–0.05, **P = 0.001–0.01, ***P = 0.0001–0.001 and ****P < 0.0001 denote statistically significant differences by two-way ANOVA. For m, n = 13–18 cells; for n, data shown are for n = 16 wells over 3 experiments. The data are presented as mean ± s.e.m. Scale bars, 25 µm (a–d, f–l). NS, not significant. Panels e,i,j created with BioRender; Anderson, P. https://BioRender.com/k5g4hqy (2026).
We next sought to evaluate the expression of endogenous β-arrestins using an orthogonal approach. To do so, we introduced the 11th β-strand of the split superfolder GFP (sfGFP; 16 amino acids), GFP11, on the C terminus of both β-arrestin 1 and 2 (ref. 30) via CRISPR–Cas9 in HEK293T cells, producing CRISPR β-arrestin-1-GFP11 and β-arrestin-2-GFP11 cell lines (Extended Data Fig. 1b). Endogenous β-arrestin can be visualized through transfection of the complementary 1–10 β-strands (GFP1–10) (Fig. 1e). We found endogenous β-arrestin 1 puncta predominantly in the cytosol (Fig. 1f). After overexpression and stimulation of β2AR or V2R, we found that β-arrestin 1 puncta translocated to the receptor (Fig. 1g,h). We also observed a similar recruitment of β-arrestin 2 puncta to the plasma membrane (Extended Data Fig. 1d).
Next we sought to evaluate endogenous β-arrestin–β-arrestin interactions. To do so, we used the CRISPR β-arrestin-GFP11 and inserted GFP1–10 on the other β-arrestin allele, producing a CRISPR β-arrestin-split GFP cell line. At baseline, we observed puncta in the cytosol and upon receptor activation (β2AR, V2R and AT1R), we observed recruitment to the plasma membrane for both β-arrestin isoforms (Extended Data Fig. 1e). To evaluate exchange of β-arrestins in these condensates, we used fluorescence recovery after photobleaching (FRAP), which revealed that β-arrestin 2-GFP11 cell line was dynamic with a recovery of about 60% (n = 6) (Extended Data Fig. 1c). Intriguingly, FRAP of β-arrestin-split GFP resulted in recovery of about 80% (n = 12), which could represent a different dynamic population of β-arrestin–β-arrestin puncta. Together, data from immunofluorescence and CRISPR cell lines demonstrate that both β-arrestin isoforms form biomolecular condensates at endogenous levels at baseline and after agonist stimulation.
β-Arrestin condensates are regulated by GPCRs
To test for the ability of β-arrestins to oligomerize when overexpressed, we compared patterns of β-arrestin labelling with monomeric and split GFP variants. Labelling of β-arrestins with enhanced GFP (eGFP) on the C terminus of β-arrestin 1 or 2 demonstrated diffuse expression in the nucleus and cytoplasm for β-arrestin 1, and in the cytoplasm alone for β-arrestin 2 due to the presence of a nuclear export sequence in β-arrestin 2 (Fig. 1i)29. To visualize potential interactions between β-arrestins, we co-expressed β-arrestin 2, which was labelled at its C terminus with the 11th β strand of split sfGFP, along with β-arrestin 2, which was labelled at its C terminus with the complementary sfGFP1–10 in HEK293T cells (Fig. 1j). Expression of β-arrestin GFP11 or β-arrestin sfGFP1–10 alone resulted in no observable signal (Extended Data Fig. 2a). With co-expression of both labelled β-arrestins, we observed the formation of puncta (Fig. 1j), consistent with an interaction between β-arrestins resulting in complementation of GFP11. These puncta localized with AP2 and clathrin, which are markers of CCPs (Fig. 1k). Notably, we did not observe visible puncta with overexpression of β-arrestin 1-eGFP and β-arrestin 2-eGFP (Extended Data Fig. 2b). Similarly, titration of split GFP-labelled β-arrestin 1 resulted in puncta near the endogenous expression of β-arrestin (Extended Data Fig. 2c,d).
We then tested the effects of agonist stimulation of the β2AR or V2R on overexpressed eGFP- or split GFP-labelled β-arrestins in HEK293T cells. These receptors display distinct patterns of β-arrestin recruitment to the receptor after agonist stimulation, where the β2AR promotes a class A pattern, characterized by plasma membrane β-arrestin redistribution but minimal internalization with the receptor to endosomes. By contrast, the V2R promotes a class B pattern that is characterized by initial β-arrestin redistribution to the plasma membrane, followed by strong localization to endosomes with the receptor31,32. Agonist stimulation of the receptors transfected with β-arrestin 2-eGFP resulted in the expected patterns at 5 and 20 min (Extended Data Fig. 3a). Cells transfected with β-arrestin-split GFP resulted in signal largely limited to the plasma membrane at both receptors at 5 min, similar to that observed with eGFP-labelled β-arrestins (Extended Data Fig. 3b). By contrast, at 20 min split-GFP-labelled β-arrestins displayed distinct patterns from eGFP-labelled β-arrestins. Although we observed some puncta at endosomes at the V2R, we still observed split GFP signal at the plasma membrane 20 minutes after agonist stimulation (Extended Data Fig. 3b). This is consistent with distinct processes being monitored by eGFP- and split GFP-labelled β-arrestin, namely redistribution and oligomerization, respectively. Together, these data are consistent with β-arrestin oligomerization being dynamically regulated by receptor activation.
To determine condensate fluidity, we next evaluated the dynamics of β-arrestin puncta with FRAP experiments (Fig. 1l,m and Supplementary Video 1). Cytosolic β-arrestin 2-eGFP showed a recovery of nearly 70% (n = 18) at 10 s. The β-arrestin 2-split GFP system showed that puncta and diffusible cytosolic pools dynamically exchange, as evidenced by similar recovery kinetics (cytosol n = 15; puncta n = 13). Quantification of the mobile fraction revealed that all three conditions showed similar patterns. We next tested whether β-arrestin oligomerization regulates the size and number of these puncta. Using previously published β-arrestin IP6 oligomerization mutants8, termed β-arrestin ΔIP6-N–C (Supplementary Fig. 1a), we found a significant decrease in both puncta intensity and number (Fig. 1n). We next tested the biophysical properties of these puncta by using 1,6-hexanediol (16HD), which disrupts π–π and hydrophobic interactions in condensates33, but can also have other off-target effects that impact cell signalling34. In cells overexpressing β-arrestin-eGFP, there was no change in the uniform distribution of β-arrestin-eGFP in the cytosol with 2.5% 16HD (Extended Data Fig. 2e). By contrast, in cells overexpressing β-arrestin-split GFP, there was a decrease in puncta intensity with 5% 16HD after 5 min (Extended Data Fig. 2e). Together, these findings are consistent with β-arrestins forming condensates that are regulated by IP6-mediated oligomerization.
Optogenetic control of β-arrestin LLPS
To dissect the components of β-arrestin sufficient for condensate formation, we leveraged an optogenetic approach to modulate condensate formation35. In short, Cry2, upon exposure to blue light, induces oligomerization, and when fused with an intrinsically disordered region (IDR), drives LLPS to form condensates (Fig. 2a). Consistent with past reports, we found that wild-type (WT) Cry2 (Cry2-mCherry) alone was insufficient in driving condensate formation (Fig. 2b,f and Supplementary Video 2)36. Fusion with the N terminus of FUS (FUSN)—a well-characterized IDR37—to Cry2WT, led to light-induced clustering in both the cytosol and the nucleus (Fig. 2c,f and Supplementary Video 3). We next fused full-length β-arrestin 1 or 2, which we termed opto-β-arrestins. For opto-β-arrestin 1, light treatment promoted condensate formation within a matter of seconds, predominantly in the cytosol (Fig. 2d,f and Supplementary Video