The ubiquitin ligase KLHL6 drives resistance to CD8+ T cell dysfunction
TL;DR
KLHL6 E3 ubiquitin ligase inhibits T cell exhaustion and mitochondrial dysfunction by degrading TOX and regulating mitochondrial fission. Its downregulation during chronic stimulation limits anti-tumor immunity, but enforced expression enhances T cell persistence and efficacy in cancer and viral models.
Key Takeaways
- •KLHL6 acts as a dual-negative regulator, reducing T cell exhaustion via TOX degradation and maintaining mitochondrial fitness by controlling fission.
- •Chronic TCR stimulation naturally downregulates KLHL6, diminishing its protective effects and contributing to T cell dysfunction in tumors.
- •Enforced KLHL6 expression in T cells improves anti-tumor and anti-viral responses, highlighting its potential as a therapeutic target in immunotherapy.
Tags
Abstract
The multifaceted dysfunction of tumour-infiltrating T cells, including exhaustion and mitochondrial dysfunction, remains a major obstacle in cancer immunotherapy1,2,3,4,5,6. Transcriptomic and epigenomic regulation of T cell dysfunction have been extensively studied7,8,9, but the role of proteostasis in regulating these obstacles remains less defined. Here we combined computational analyses of atlases of T cell exhaustion and mitochondrial fitness with performed targeted in vivo CRISPR screens, which identified the E3 ubiquitin ligase KLHL6 as a dual-negative regulator of both T cell exhaustion and mitochondrial dysfunction. Mechanistically, KLHL6 expression promoted TOX poly-ubiquitination and subsequent proteasomal degradation, thereby attenuating the transition of progenitor exhausted T cells towards terminal exhaustion. Simultaneously, KLHL6 maintained mitochondrial fitness by constraining the excessive mitochondrial fission that occurs during chronic T cell receptor stimulation by means of post-translational regulation of the PGAM5–Drp1 axis. However, KLHL6 is naturally downregulated by T cell receptor ligation, mitigating its potentially beneficial ubiquitin ligase activities during exposure to chronic stimulation. Enforcing KLHL6 expression in T cells markedly improved efficacy and long-term persistence against tumours and during viral infections in vivo. These findings uncover KLHL6 as a multifunctional, clinically actionable target for cancer immunotherapy, and highlight the potential of modulating proteostasis and ubiquitin modification to improve immunotherapy.
Main
Durable responses to T-cell-based cancer immunotherapies remain limited in a substantial fraction of patients10,11. A major barrier to efficacy is T cell dysfunction within the tumour microenvironment (TME), driven in part by exhaustion, mitochondrial dysfunction and various immunosuppressive factors12,13. Despite extensive transcriptomic and epigenomic insights into T cell dysfunction7,8,9, our understanding of how post-translational modifications affect proteostasis to contribute to regulating this multifaceted process is still limited. Ubiquitin ligases have important roles in controlling protein degradation and homeostasis14, thereby modulating diverse biological events, including T cell development, activation and trafficking15,16,17. The multifunctional nature of these ligases, which can target diverse substrates for degradation, offers unique opportunities to address many obstacles of anti-tumour T cell responses simultaneously. However, the fundamental roles of the diverse universe of ubiquitin ligases, particularly in anti-tumour T cell biology, remain incompletely understood, limiting potential modulation of ubiquitin ligase activity as a therapeutic strategy in cancer immunotherapy. In this study, we integrated computational analyses with targeted in vivo CRISPR screens, and identified the E3 ubiquitin ligase KLHL6 as a dual-negative regulator of T cell exhaustion and mitochondrial dysfunction during chronic antigen stimulation. Enforced expression of KLHL6 in adoptively transferred T cells improved anti-tumour and anti-viral efficacy. The findings highlight the crucial role of ubiquitin modifications in dictating T cell fate and function, and implicate KLHL6 as a promising clinically actionable multifunctional target to enhance cancer immunotherapy.
CRISPR screen for E3 ligases in T cells
To elucidate general mechanisms of T cell dysfunction, we analysed 136 bulk RNA sequencing (RNA-seq) samples of CD8+ T cells from eight independent infection and cancer studies to define the exhaustion transcriptomic landscape (Fig. 1a, Extended Data Fig. 1a and Supplementary Table 1). Information-theoretic analyses distilled transcriptional variation across thousands of genes into two core modules (modules 1 and 2), each comprising opposing gene sets (Extended Data Fig. 1a and Supplementary Table 2). These two modules recapitulated the global transcriptome of CD8+ T cells under chronic (tumour and/or infection) and acute (infection) conditions (Extended Data Fig. 1a), capturing the principal programs underlying exhaustion. Module 1 positively enriched genes were low in naive and memory T cells but high in early and late exhausted T (Tex) cells, whereas negatively enriched genes showed the inverse trend. Module 2 positively enriched genes were selectively elevated in late exhausted cells, and negatively enriched genes were higher in naive and early exhausted states (Extended Data Fig. 1a and Supplementary Table 2). Gene set enrichment analysis (GSEA) revealed functional distinctions: module 1 genes associated with chronic stimulation, exhaustion and differentiation from naive and/or memory states, whereas module 2 genes related to proliferation, self-renewal and progenitor exhausted T (Tpex) cell features (Extended Data Fig. 1b,c and Supplementary Table 3). Projecting time-course datasets of antigen-specific CD8+ T cells (acute, Listeria or chronic, LCMV-Clone 13/tumour) onto a two-module map confirmed module 1 effectively separated acute from chronic antigen exposure, and module 2 tracked early-to-late exhaustion progression8,18 (Fig. 1b,c). Projecting independent time-series transcriptomes of CD8+ T cells in acute and chronic LCMV infection19 onto the same map revealed a coherent temporal trajectory of CD8+ T cell differentiation, with early samples (day 6–8) from both infections clustering together, consistent with early precursor T cells adapting to both acute and chronic infection20,21 (Extended Data Fig. 1d,e). These findings support the biological relevance of the two gene modules in capturing dynamic trajectories of CD8+ T cell differentiation across several physiological contexts.
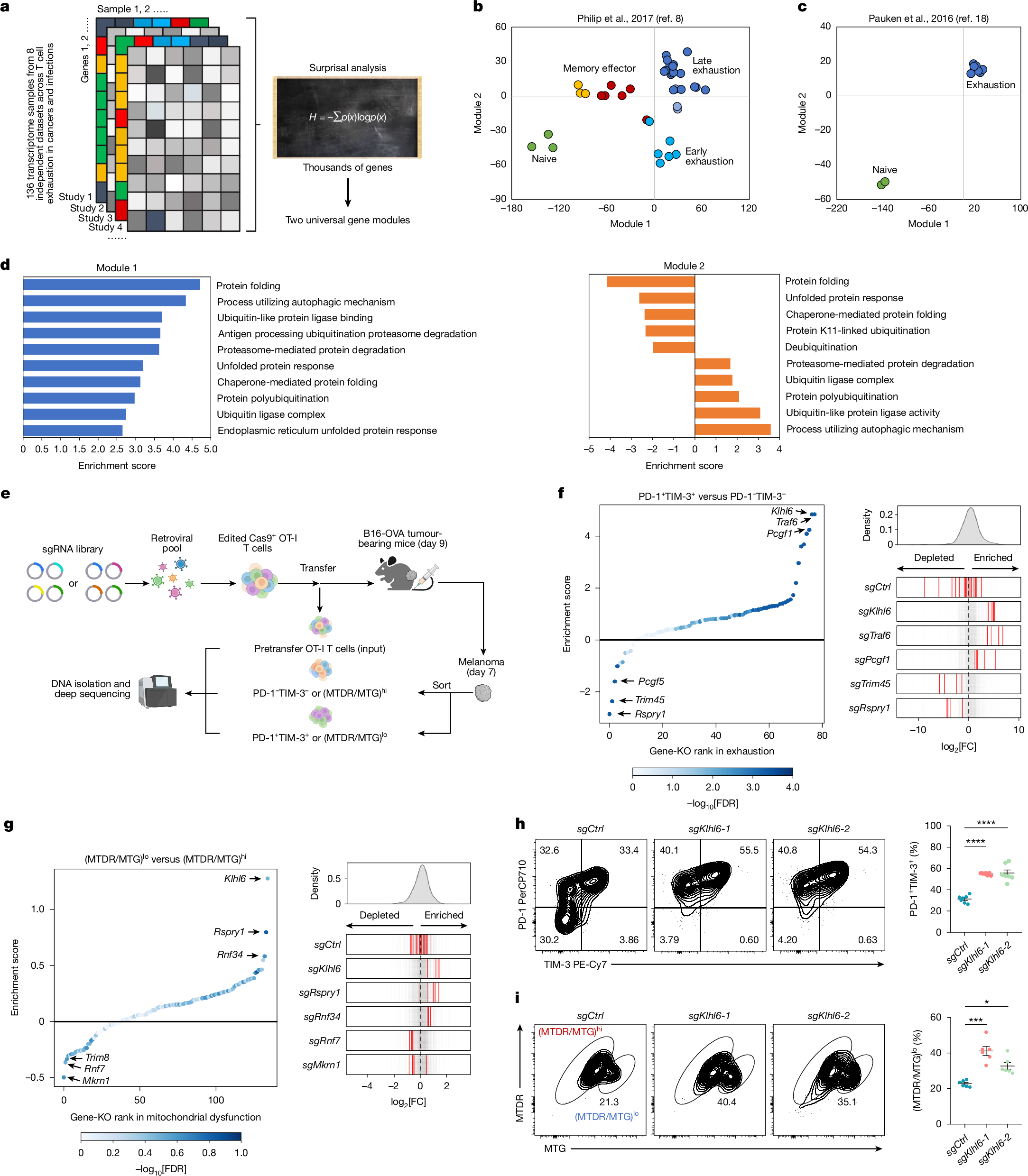
a, Schematic illustration of workflow for computational analysis of RNA-seq atlases of T cell exhaustion. b,c, Samples from two published datasets (GSE89307, GSE86881) were projected onto a two-dimensional map defined by computationally derived gene modules. b, Antigen-specific TCR-transgenic T cells collected across acute Listeria infection and tumour progression at matched time points following adoptive transfer. c, Antigen-specific naive and chronically exhausted T cells isolated during late-stage LCMV infection. d, GSEA enrichment of proteostasis-associated pathways in modules 1 and 2. e, Experimental schematic of CRISPR screen for E3 ligases that regulate T cell exhaustion and mitochondrial function. f,g, Rank plots (left) of gene-level enrichment scores in exhausted versus non-exhausted T cells (f) and in T cell populations with dysfunctional versus functional mitochondria (g). The CRISPR enrichment scores (log2 fold change of PD-1+TIM-3+ versus PD-1−TIM-3− or (MTDR/MTG)lo versus (MTDR/MTG)hi) were determined by comparing the indicated subsets for target genes from Cas9+sgRNA (mCherry)+ cells isolated from tumours on day 7 after ACT. The x axis shows targeted genes; the y axis shows the CRISPR enrichment score of each targeted gene; the dot colour represents false discovery rate (FDR). Distribution of several top-hit sgRNAs (right). Axis represents log2 fold change (FC). The histogram shows distribution of all sgRNAs. Red bars represent targeted sgRNAs, grey bars represent all other sgRNAs. h,i, Representative plots (left) and quantification (right) of the proportions of PD-1+TIM-3+ (h, n = 7 mice) and (MTDR/MTG)lo (i, n = 6 mice) TILs in sgCtrl-transduced or sgKlhl6-transduced Cas9+ OT-I T cells from B16-OVA tumours obtained on day 7 after ACT. Diagram in a created in Biorender. Li, G. (2025) https://BioRender.com/d5c767f. Diagram in e created in BioRender. Li, G. (2025) https://BioRender.com/dw0yfsp. Data are presented as mean ± s.e.m. Statistical analyses were determined by two-way ANOVA with Tukey’s multiple-comparisons test (h,i). *P < 0.05, ***P < 0.001 and ****P < 0.0001.
Notably, we observed significant enrichment of proteostasis-related pathways in modules 1 and 2, particularly the ubiquitin–proteasome system (UPS) (Fig. 1d and Supplementary Table 3). Given the limited understanding of proteostasis in T cell dysfunction, we focused on the UPS pathway. Hierarchical clustering of E3 ligase-related genes from OT-I T cells isolated from B16-OVA tumours identified two patterns, with one upregulated in single positive (SP, PD-1+TIM-3−) and double-positive (DP, PD-1+TIM-3+) tumour-infiltrating T cells (TILs) and the other downregulated (Extended Data Fig. 1f–h), underscoring potential roles of E3 ligase-mediated proteostasis in shaping exhausted T cell differentiation. Given the link between mitochondrial dysfunction and exhaustion under TME metabolic stress3,4,5,6, we analysed mitochondrial-associated genes and pathways in human and mouse TILs6,22, again revealing strong associations between UPS and mitochondrial function (Extended Data Fig. 1i,j). To prioritize functional candidates, we selected 78 E3 ligase-related genes negatively enriched in both exhaustion modules and 133 E3 ligase candidates positively associated with mitochondrial function for two parallel in vivo CRISPR screens targeting exhaustion and mitochondrial fitness (Extended Data Fig. 1k,l and Supplementary Table 4).
Using an established in vivo CRISPR-screening system23, single-guide RNA (sgRNA)-transduced Cas9+ OT-I T cells were transferred into B16-OVA tumour-bearing mice (Fig. 1e). Seven days after adoptive cell transfer (ACT), PD-1+TIM-3+ versus PD-1−TIM-3− cells (exhaustion screen) and (MitoTracker Deep Red (MTDR, mitochondrial membrane potential)/MitoTracker Green (MTG, mitochondrial mass))lo versus (MTDR/MTG)hi cells (mitochondrial dysfunctional versus functional screen) were isolated for sgRNA enrichment (Fig. 1e and Supplementary Table 5). Exhaustion screen identified positive and negative regulators including Klhl6, Traf6, Pcgf1, Trim45 and Rspry1 (Fig. 1f), with Traf6 known to promote CD8+ T cell memory and anti-tumour responses24,25 but underexplored in exhaustion. The mitochondrial screen revealed several established regulators such as Rnf34, Mkrn1 and Rnf7 (refs. 26,27,28), not previously studied in T cells, and new regulators such as Klhl6, Trim8 and Rspry1 (Fig. 1g). Integrating both screens, KLHL6 emerged as the top-ranked E3 ligase regulating both T cell exhaustion and mitochondrial fitness (Fig. 1f,g). In vivo validation with two Klhl6-targeting sgRNAs confirmed Klhl6 deletion reduced OT-I cell accumulation in the TME and increased frequencies of exhausted (PD-1+TIM-3+ or LAG-3+TIM-3+) and mitochondrially depolarized (MTDR/MTG)lo CD8+ TILs (Fig. 1h,i and Extended Data Fig. 2a–e). Further validation confirmed Pcgf1 or Traf6 deletion increased exhausted PD-1+TIM-3+CD8+ TILs, whereas Rspry1 knockout (KO) reduced exhaustion, consistent with screening results (Extended Data Fig. 2f). Phenotypic assessments of mitochondrial regulators aligned with screen results (Extended Data Fig. 2g). These findings strongly implicate KLHL6 as a critical regulator of both T cell exhaustion and mitochondrial function.
KLHL6 deficiency impairs T cell function
The ubiquitinase KLHL6, a Cullin3-RING E3 complex component, is known to regulate B cell differentiation29,30. To investigate its role in T cell immunity, we generated KLHL6-deficient (Klhl6−/−) mice and evaluated immunological characteristics (Extended Data Fig. 2h,i). As reported in ref. 30, KLHL6 deficiency impaired mature B cell formation but had no great effect on T cell development, peripheral homeostasis or activation, and only modestly reduced proliferation in vitro (Extended Data Fig. 2j–o). To assess the impact on anti-tumour T cell responses in a mouse B16-OVA melanoma model (Extended Data Fig. 2p), we crossed Klhl6−/− mice with OT-I mice (Klhl6−/− OT-I mice). By day 14, Klhl6−/− OT-I cells showed weaker tumour control than wild-type (WT) cells (Fig. 2a), with reduced accumulation in tumour, draining lymph node (dLN) and spleen (Fig. 2b and Extended Data Fig. 2q). Klhl6−/− CD8+ TILs showed increased PD-1+TIM-3+ and reduced PD-1−TIM-3− and PD-1+TIM-3− populations (Fig. 2c), elevated PD-1, TIM-3, LAG-3 and TOX expression (Extended Data Fig. 2r,s), and diminished tumour necrosis factor (TNF) and IFNγ production (Fig. 2d). Mice receiving Klhl6−/− OT-I cells had poorer survival (Extended Data Fig. 2t). To minimize inter-tumour variability, equal WT and Klhl6−/− OT-I T cells were cotransferred into the same tumour-bearing hosts (Fig. 2e). Klhl6−/− cells were consistently significantly decreased in tumour, dLN and spleen (Fig. 2f), and showed elevated PD-1, TIM-3 and TOX expression within tumours (Fig. 2g–i). KLHL6 deletion induced broad transcriptional changes (Extended Data Fig. 3a), including upregulation of exhaustion-related genes and downregulation of stemness-associated genes (Fig. 2j and Extended Data Fig. 3b). Gene Ontology analysis showed enriched oxidative stress-induced senescence, transforming growth factor-β (TGFβ) signalling and G2 cell-cycle arrest pathways in KLHL6-deficient cells (Extended Data Fig. 3c). GSEA revealed reduced memory and effector signatures but increased apoptosis and cell-cycle arrest signatures in Klhl6−/−CD8+ TILs (Extended Data Fig. 3d,e). Collectively, KLHL6 loss drove CD8+ T cells towards exhaustion and dysfunction.
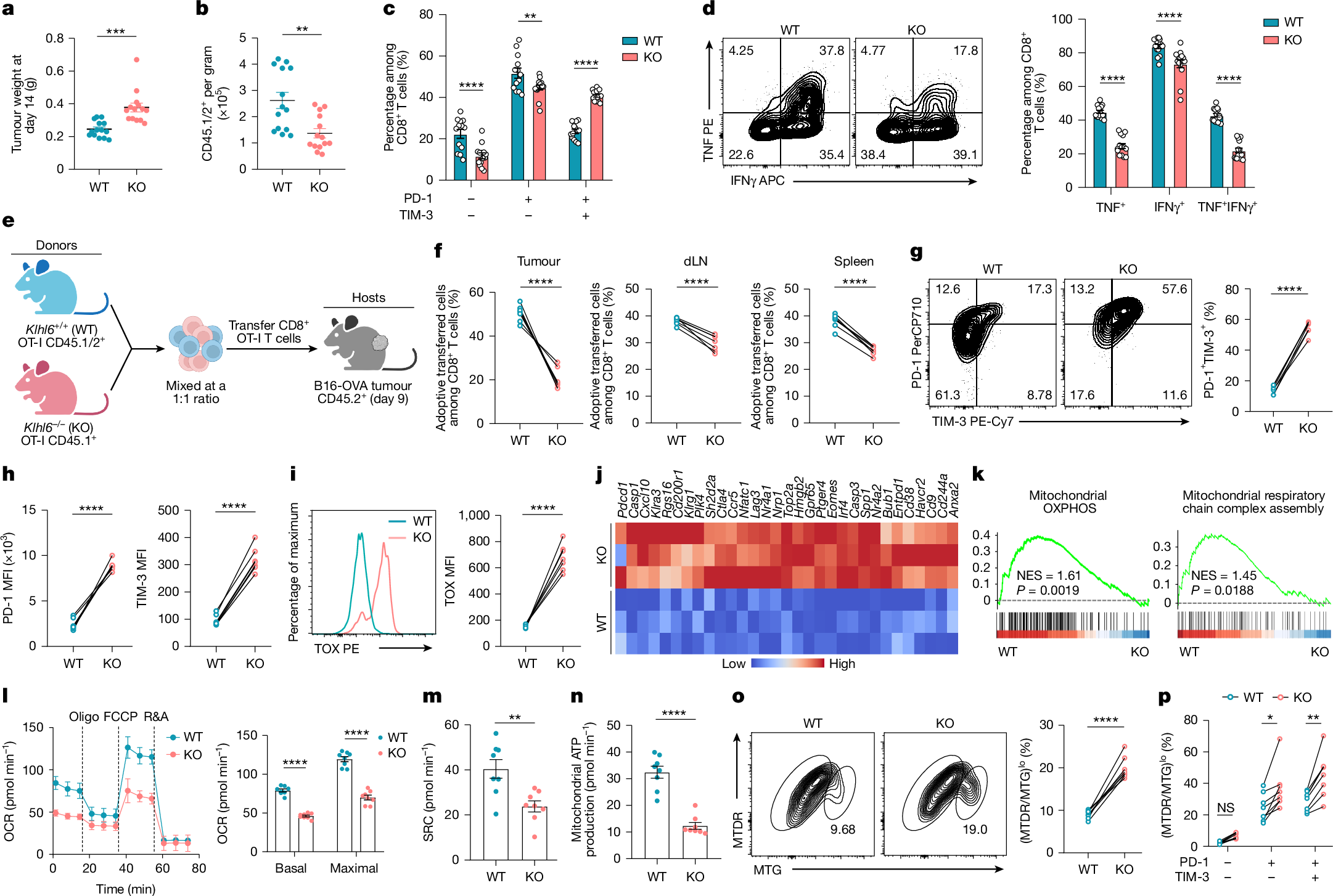
a,b, CD45.2+ C57BL/6N mice were subcutaneously injected with B16-OVA melanoma cells and, 9 days later, treated by ACT with 3 × 106 CD45.1/2+ Klhl6+/+ (WT) or Klhl6−/− (KO) OT-I T cells (n = 14 mice). Tumour weights (a) and the numbers of transferred OT-I T cells (b) were assessed at day 14 post-ACT. c, Percentages of PD-1−TIM-3−, PD-1+TIM-3− and PD-1+TIM-3+ subsets among CD45.1/2+ TILs (n = 14 mice). d, Cytokine production (TNF and IFNγ) in transferred CD45.1/2+ TILs was determined (n = 14 mice). e–i, For cotransfer experiments, CD45.1/2+ WT and CD45.1+ KO OT-I T cells were mixed at a 1:1 ratio and then adoptively transferred into tumour-bearing CD45.2+ mice 9 days after tumour inoculation, and the mice were euthanized for analysis at day 14 after ACT (n = 7 mice). Experimental design (e), the proportions of WT and KO OT-I cells in tumour, dLN and spleen (f), the percentages of PD-1+TIM-3+ population (g) and the expression levels of PD-1 and TIM-3 (h) and TOX (i) in cotransferred WT and KO TILs. j, Heat map of exhaustion-associated genes in WT and KO TILs at day 14 post-ACT (n = 3 independent samples). k, GSEA plots of signatures of mitochondrial OXPHOS and mitochondrial respiratory chain complex assembly in WT versus KO TILs (n = 3 independent samples). GSEA was performed using a one-sided, permutation-based modified K–S test with adjustments for multiple comparisons. l–n, Cotransferred WT and KO TILs were sorted for Seahorse assays on day 14 post-ACT. OCR (l), SRC (m) and mitochondrial ATP production (n) were measured (n = 8 independent tests; 20 mice). o,p, The frequencies of (MTDR/MTG)lo subsets in transferred WT and KO OT-I TILs (o) and their distribution among exhausted (PD-1+TIM-3− and PD-1+TIM-3+) and non-exhausted (PD-1−TIM-3−) populations (p) at day 14 after ACT (n = 8 mice). Diagram in e created in BioRender. Li, G. (2025) https://BioRender.com/5se3g09. Data are presented as mean ± s.e.m. Statistical analyses were determined by unpaired two-tailed Student’s t-test (a,b,f–i,m–o) or two-way ANOVA with Sidak’s multiple-comparisons test (c,d,l,p). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; NS, not significant. FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; MFI, mean fluorescence intensity; NES, normalized enrichment score; R&A, rotenone and antimycin A.
GSEA also showed reduced OXPHOS and respiratory chain activity in Klhl6−/− T cells (Fig. 2k and Extended Data Fig. 3f). MTG and tetramethylrhodamine ethyl ester (TMRE) (membrane potential) staining revealed a markedly reduced TMRE/MTG ratio, indicating impaired mitochondrial activity (Extended Data Fig. 3g). Metabolic analyses demonstrated reduced oxygen consumption rate (OCR), spare respiratory capacity (SRC) and ATP production but elevated glycolytic proton efflux rate (glycoPER) in Klhl6−/− T cells (Extended Data Fig. 3h–k). Similarly, KLHL6-deficient TILs showed diminished mitochondrial function, increased glycoPER and a higher frequency of (MTDR/MTG)lo depolarized mitochondria compared with WT cells6,31 (Fig. 2l–o and Extended Data Fig. 3l–n). Exhausted TIL subsets (PD-1+TIM-3−, PD-1+TIM-3+) harboured more depolarized mitochondria than non-exhausted (PD-1−TIM-3−), and KLHL6 deficiency further exacerbated depolarization in exhausted TILs (Fig. 2p), underscoring a positive regulatory role maintaining mitochondrial fitness.
KLHL6 expression profiling of OT-I subpopulations from spleen and tumour (PD-1−TIM-3−, PD-1+TIM-3− and PD-1+TIM-3+) revealed Klhl6 expression highest in splenic T cells followed by PD-1−TIM-3− cells compared with the other tumour-reactive subsets (Extended Data Fig. 4a). T cell receptor (TCR) signalling downregulated KLHL6 protein expression in vitro (Extended Data Fig. 4b,c). Time-series analysis showed transient decline-recovery of KLHL6 expression following TCR stimulation but sustained repression on repeated stimulation (Extended Data Fig. 4d,e). Transcriptomic analysis of CD8+ T cells in acute and chronic infections or tumours8,19, confirmed persistent KLHL6 downregulation in chronic but transient reduction in acute contexts (Extended Data Fig. 4f,g). These findings were validated with an ex vivo time-course analysis of KLHL6 messenger RNA (mRNA) and protein levels during acute (Armstrong) and