Mechanism of age-related accumulation of mtDNA mutations in human blood
Abstract
Accumulation of mutant mitochondrial DNA (mtDNA) heteroplasmy is among the strongest signatures of ageing1. Here we investigated the underlying mechanism by calling mtDNA sequence, mtDNA abundance and mtDNA heteroplasmic variants in human blood using whole-genome sequences from approximately 750,000 individuals. We observed that mtDNA single-nucleotide variants (mtSNVs) accumulate sharply at age 60 years, occur at low levels of heteroplasmy, exhibit little evidence of positive selection and are likely to be predominantly neutral. The mutational spectrum of mtSNVs does not reflect oxidative lesions, as is commonly invoked, but is more consistent with mtDNA replication errors. To understand why mtSNVs become detectable with age, we performed a genome-wide association study for heteroplasmic mtSNV burden, identifying germline variants near TERT, TCL1A and SMC4, all of which have been linked to clonal haematopoiesis (CH)2. Rare-variant analysis also showed that high mtSNV burden is associated with mutations in numerous CH driver genes. These genetic associations persisted even after exclusion of individuals with known CH driver mutations. Our results support a model in which ‘cryptic’ mtDNA mutations initially arise randomly as replication errors but are undetectable in bulk. They then become apparent only through age-related expansion of cellular clones in blood. We propose that the high copy number and mutation rate of mtDNA make it a sensitive blood-based marker of somatic mosaicism due to CH. Our work mechanistically unifies three prominent signatures of ageing: common germline variants in TERT, CH and observed accrual of mtDNA mutations.
Similar content being viewed by others

Nuclear genetic control of mtDNA copy number and heteroplasmy in humans


Single-cell multi-omic analysis of mitochondrial mutational mosaicism and dynamics
Main
Mitochondrial DNA (mtDNA) heteroplasmy arises when a cell or tissue contains a mixture of two or more different mtDNA alleles. Heteroplasmy dynamics tend to be complex, varying across generations, during development, in disease and with ageing. Historically, most studies of mtDNA heteroplasmy in humans have focused on rare, maternally transmitted disorders, which are typically driven by loss-of-function mtDNA mutations at high levels of heteroplasmy. However, there is growing evidence that low levels of mtDNA heteroplasmic variants are found in nearly all humans3. Using biobank-scale genomics, we previously reported that nearly everyone harbours two different classes of such variants in blood4. ‘Length heteroplasmies’ (insertion/deletion (indel) mutations within polypyrimidine tracts) do not accumulate with age, tend to be maternally transmitted and, once inherited, exhibit levels of heteroplasmy under nuclear genetic control. The associated nuclear DNA (nucDNA) loci tend to implicate mitochondria-localized proteins with established roles in mtDNA replication and maintenance. By contrast, heteroplasmic mtSNVs tend not to be inherited but rather seem to be somatic in origin and accumulate with age4.
The mechanism of this age-related accrual of heteroplasmic mtSNVs in blood is unknown. Classically, oxidative damage to mtDNA from reactive oxygen species has been invoked as a part of a ‘vicious cycle’ in which mtDNA mutations lead to further generation of reactive oxygen species and more mutations5,6,7. More recent studies of ageing brains8 and tumour samples9 have questioned the role of oxidative damage10 in mutation generation. Once individual mutations arise, it is unclear how they become abundant enough to detect.
Here we investigated why mtSNVs accumulate with age in blood. We report an analysis of mtDNA using a callset of approximately 750,000 individuals across the UK Biobank (UKB) and All of Us (AoU). We resolved the mutational spectrum of age-accumulating blood mtDNA and then performed a genome-wide association study (GWAS) for the burden of mtSNVs to identify mechanisms of control by rare and common germline nuclear genetic variants. Unexpectedly, the loci we identified were related not to mitochondrial homeostasis but rather to CH. Our analyses support a model in which individual blood cells randomly accumulate low levels of neutral mtDNA variation (that is, cryptic mutations), probably owing to replication-related errors, that are not detectable in bulk. However, with age-related expansion of individual cellular clones (by means of CH), these low-level cryptic variants become detectable and give rise to the observed accumulation with age.
mtDNA sequences from 736,038 individuals
We applied mtSwirl4, a pipeline for calling mtDNA abundance and heteroplasmic variation (Methods), to whole-genome sequencing (WGS) data from 736,038 diverse individuals in AoU and UKB, representing an approximately 3× increase in sample size compared with ref. 4. mtSwirl reconstructs each individual’s mtDNA to serve as a ‘self-reference’ against which low levels of heteroplasmic variants can be more accurately called4. After quality control, we called a total of 19,051,526 variants (Methods) from 620,385 individuals. Of the 1,151,297 heteroplasmic variants (Supplementary Note 1), 754,108 were indels and 397,189 were SNVs (Extended Data Fig. 1).
To benchmark our callset, we performed GWAS of mtDNA copy number (mtCN) and individual common heteroplasmic variants and compared them with the results of our previous study4. GWAS for blood-composition-adjusted mtCN in UKB (mtCNadj; Supplementary Note 2) across 398,250 individuals identified 107 loci (Extended Data Fig. 2c and Methods), many of which were corroborated by fine-mapping and gene-based rare-variant testing (Extended Data Fig. 2d,e). We replicated 44 of 46 loci from our previous mtCNadj GWAS4 (n = 163,372) at genome-wide significance (GWS) and discovered 63 further associations (for instance near LIG3 and TBRG4). As in previous work4,11, adjustment for blood cell composition eliminated or reversed the direction of most associations between reduced mtCN and increased rates of age-related diseases (Extended Data Fig. 2f). GWAS for heteroplasmy of each of 78 common mtDNA variants, most of which were indels (Extended Data Fig. 1), identified 163 associations across 60 nuclear loci (Extended Data Fig. 3a). We replicated virtually all previously identified associations4 and identified many more hits. For example, we report associations between chrM:302:A:AC (the most common heteroplasmy in humans) and nucDNA variants near TSFM, TOP3A, PANK1 and UQCRC1; UQCRC1 is supported by a fine-mapped missense variant (Extended Data Fig. 3b). Across mtDNA sites, DGUOK and PNP were associated with most heteroplasmies (N < 42,000) except for chrM:302 (n = 244,879) (Extended Data Fig. 3a–f), implying differences in nuclear control of heteroplasmy across mtDNA sites beyond the effects of power (Supplementary Note 3). Recessive GWAS revealed no new loci (Extended Data Fig. 3g,h).
Heteroplasmic mtSNVs reflect replication error
Focusing on heteroplasmic mtSNVs in AoU, we observed mutation accrual particularly after the age of 60 years (Fig. 1). We found a similar mtSNV accumulation in UKB (Extended Data Fig. 4a). This accumulation occurred regardless of smoking status; however, heteroplasmic SNVs were more abundant in smokers, as described previously12 (Extended Data Fig. 4b).
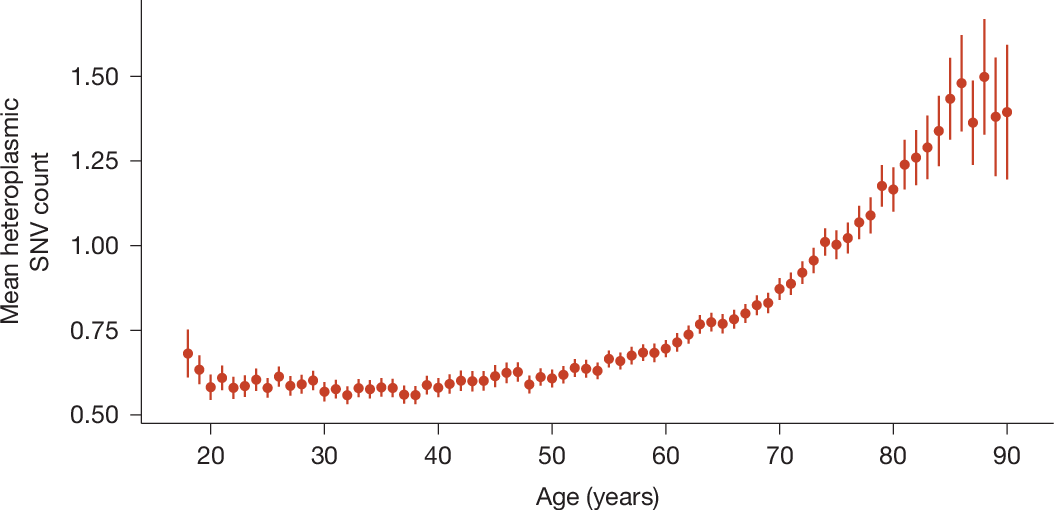
Points represent means, error bars represent 95% confidence intervals. See Extended Data Fig. 4a for the corresponding analysis in UKB.
We first visualized mtDNA mutations by variant type and strand. mtDNA contains a ‘heavy strand’ with more purines and a ‘light strand’ with more pyrimidines. Replication begins on the light strand at the origin of replication (Ori). We excluded Ori, as it has been shown to have a different mutational spectrum from the coding mtDNA9. Outside Ori, most variants were transitions (Fig. 2a,b and Extended Data Fig. 5a,b) with a striking strand bias: C>T and A>G variants occurred far more frequently on the heavy strand than on the light strand.
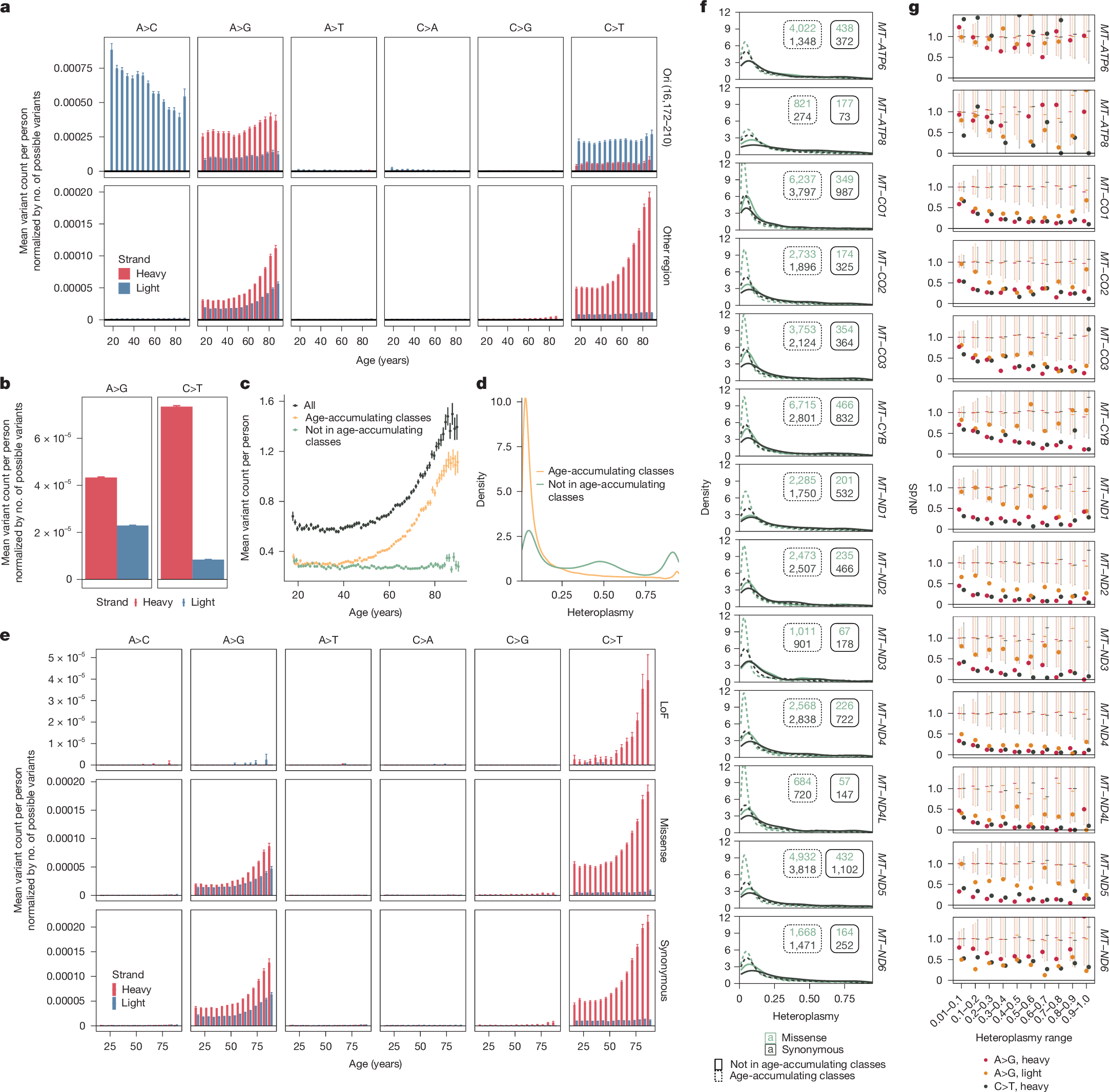
a, Normalized mean mtSNV count as a function of variant location, strand, class and age. b, Normalized mean mtSNV count as a function of strand and variant class. c, Mean mtSNV count as a function of age for all variants, age-accumulating class variants (C>T heavy and A>G heavy and light) and other variants. d, Heteroplasmy distribution across variants and individuals for age-accumulating classes only and all variants. e, Normalized mean mtSNV count for coding variants as a function of strand, class, age and consequence. f, Heteroplasmy distributions for missense (green) versus synonymous (black) variants within mtDNA protein coding genes, stratified by age-accumulating class (dashed versus solid). Insets are corresponding sample sizes. g, dN/dS estimates (dots) as a function of heteroplasmy, gene and mutation class for age-accumulating class variants in mtDNA protein coding genes. Dashes indicate the median dN/dS of null draws; error bars are 95% confidence intervals under null, computed non-parametrically as the 2.5–97.5% range of null draws. In a–c and e, error bars indicate ±1 s.e. For a, c and e, total n = 236,749 individuals. For b and g, total n = 237,500 individuals. All panels in this figure use AoU data; see Extended Data Fig. 5 for UKB.
Classically, mutations in mtDNA have been thought to arise owing to oxidative stress7; however, growing evidence over the past decade8,9,13 has suggested that replication-linked errors may better explain heavy-strand-biased transition mutations. Under the strand displacement model, once replication of the light strand begins at Ori, the non-template heavy strand persists in a single-stranded state until replication progresses through approximately two-thirds of the molecule14. A bias towards heavy strand C>T and A>G mtSNVs has been observed in human brain and tumour samples and attributed to deamination from the protracted time that the heavy strand spends in a single-stranded state9,15. By contrast, oxidative damage predominantly produces C>A transversions due to guanosine oxidation16,17,18. Consistent with findings in human brain and tumours8,9, our observation of heavy-strand-biased C>T and A>G mutations with little C>A in blood suggests replication-related errors with little oxidative damage.
Although mechanisms of mtDNA and nucDNA replication are distinct, we briefly explored whether our observed mtDNA mutation spectrum had parallels with nucDNA. Our observed pattern of mtSNVs was most correlated with known nuclear single-base substitution signatures attributed to nucDNA damage repair defects (for instance, mismatch repair) (Extended Data Figs. 6a,b and 7a,b). On the other hand, nuclear mutation signatures attributed to oxidative damage19,20,21 were not correlated with our observed mtDNA mutational signature.
Only C>T mutations (on the heavy strand) and A>G mutations (on both strands) accumulated with age (Fig. 2a and Extended Data Fig. 5a), whereas heteroplasmic SNVs outside these classes did not (Fig. 2c and Extended Data Fig. 5c). In contrast to A>G and C>T heavy strand mutations, for which all trinucleotide contexts showed age accumulation (Extended Data Figs. 6c,d and 7c,d), only a subset of A>G light strand contexts showed age accumulation. A contributing mechanism may be polymerase error, which can produce transitions22 and could be context-specific and strand independent.
We previously showed that mtSNV heteroplasmy tends to be somatic and not maternally transmitted4. Here we wanted to know whether this observation extended to age-accumulating class variants (that is, A>G on both strands and C>T on heavy strand). SNVs with high heteroplasmy (higher than 0.2) showed a greater than 75% chance of being found in both siblings, whereas low-heteroplasmy (0.2 or lower) variants had a less than 30% chance of being shared between siblings (Extended Data Fig. 4c). To control for the possibility that sibling-sharing could have been present but below our detection threshold, we repeated this analysis for chrM:302:A:AC, which tends to be maternally inherited4; we found that more than 60% of variants at heteroplasmy less than 0.2 showed sibling-sharing. Most age-accumulating class variants had heteroplasmy less than 0.2, substantially less than that of non-age-accumulating class variants (Fig. 2d and Extended Data Figs. 4d,e and 5d). Overall, these results indicate that age-accumulating class mtSNVs may have lower heteroplasmy and are more likely to be somatic than non-age-accumulating class variants.
Age-accumulating mtSNVs appear neutral
We next sought to determine whether positive selection shaped the observed pattern of heteroplasmic mtSNV accrual. We observed a similar strand bias among age-accumulating class variants regardless of whether the variant was missense or synonymous (Fig. 2e and Extended Data Fig. 5e), arguing against selection as a central driver. Missense variants had left-shifted heteroplasmy distributions relative to synonymous variants for all genes, suggesting selection against coding variants at higher levels of heteroplasmy (Fig. 2f and Extended Data Fig. 5f). A metric traditionally used to quantify the degree of selection acting on variation is the normalized ratio between non-synonymous and synonymous variation (dN/dS), where a ratio less than 1 suggests negative selection, approximately 1 suggests neutrality and greater than 1 suggests positive selection. In mtDNA, this metric must account for the underlying mutational process (which is strand- and mutation-class-specific) and codon composition. Accordingly, we used two methods to estimate and interpret dN/dS: first, a non-parametric approach recently used in mtDNA23; second, a parametric approach accounting for strand and mutation class24 (Methods). We found that dN/dS among age-accumulating variants was closest to neutral at low heteroplasmy and fell as heteroplasmy rose in all genes except MT-ATP6, MT-ATP8 and MT-ND6 (Fig. 2g and Extended Data Figs. 5g, 6e and 7e). These results indicate that purifying selection against deleterious alleles may increase with higher levels of heteroplasmy, consistent with the ‘heteroplasmy threshold effect’ in which high levels of heteroplasmy are required before phenotypic effects of the mutation are observed