Transcription factor codes patterning neuronal groundplans of the cerebrum
Abstract
Brain regions that regulate motivated behaviours, including the vertebrate hypothalamus and arthropod cerebrum, house bespoke neural circuits dedicated to perceptual and internal regulation of many behavioural states1,2. These circuits are built to purpose from complex sets of cell types whose patterning has been challenging to elucidate. Here we developed methods in Drosophila melanogaster to embed well-studied neurons that regulate mating in the transcriptional contexts of the neuronal lineages that generate them3,4,5. By comparing transcription within and between lineages, we identified a large set of transcription factors expressed in complex combinations that delineate cerebral hemilineages—classes of postmitotic neurons born from the same stem cell and sharing Notch status6,7. Hemilineages comprise the major anatomic classes in the cerebrum8,9,10 and these transcription factors are required to generate their gross features. We show that subtypes of the same hemilineage can provide a common computational module to circuits regulating different drives, and identify an orthogonal set of transcription factors that stratify hemilineage subtypes of differing birth order. Our findings suggest that distinct sets of transcription factors operate in a hierarchical system to build, diversify and sexually differentiate lineally related neurons that compose motivated behaviour circuits. By linking developmental patterning to separable transcriptional axes that produce gross versus fine aspects of information flow, we provide a logical framework for cerebral control of diverse drives.
Similar content being viewed by others

An atlas of gene regulatory elements in adult mouse cerebrum

A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain

Decoding gene regulation in the fly brain
Main
Motivated behaviours, such as mating, feeding or aggression, are regulated by neurons that each perform singular information-processing functions1,2. These neurons are organized into unique feed-forward circuits in vertebrate subcortical nuclei and the arthropod cerebrum. The genome contains the information necessary to assign meaning to sensory inputs and to organize relationships among different drives into motivated behaviour circuits. Yet the immense diversity of contributing neurons has made their patterning too difficult to address. To explain the anatomic and functional principles that structure these circuits and states, we need to describe the logical principles and molecular mechanisms through which their constituent neurons are patterned during development.
Four logical axes diversify neuronal cell types. They are used distinctly across brain regions in organisms, and in different kinds of neurogenesis programs across clades11,12,13: neural stem cells are (1) spatially differentiated and (2) progress through temporal expression windows as they divide asymmetrically; differentiating daughter cells can be further separated by (3) a Notch switch; and (4) sex further differentiates a subset of cell types.
In the arthropod cerebrum, approximately 200 anatomic units called hemilineages form an information highway system traversing brain regions, as their constituent neurons share neurite outgrowth tracts, gross morphology and, often, neurotransmitter systems8,9,10,14,15,16,17. In adult Drosophila melanogaster (D. melanogaster), cerebral hemilineages—like those of the ventral nerve cord (VNC)—are composed of the NotchON or NotchOFF cousins from the same neuroblast7,8,9,10,18,19. Within hemilineage ground plans, individual cells have identifiable differences in fine axonal and dendritic anatomy and connectivity, linked to their birth order15,18,20,21. Although lineages of nerve cords, visual systems or cortices repeat across segments or columns, the unique functions of the cerebrum are produced by mostly singular lineages. Thus cerebral cell types, defined at the level of anatomy, connectivity or circuit function, are often specific birth-order cohorts from within one hemilineage. This singularity is what makes the complex circuit computations of the cerebrum possible; at the same time, the rarity of these neuroblasts and their daughter cells has impeded efforts to describe the transcriptional mechanisms that link neurogenic patterning to the formation of circuit architecture.
Here we break this impasse by starting with the unusually well-understood circuit that regulates male mating in D. melanogaster. This circuit is built from over 60 populations of neurons that arise as subtypes of defined lineages and that are sexually differentiated by the Fruitless and/or Doublesex transcription factors (TFs)3,4,10,18,22,23. We transcriptionally characterized neuronal subtypes in the mating circuit in the context of their lineages and identified groups of TFs that play separable logical roles in diversifying hemilineage ground plans, versus birth-order-associated subtypes within hemilineages. Focusing on a hemilineage that generates sexually differentiated neurons that gate mating, we found that mutation of hemilineage TFs disrupted general morphological features; that manipulation of birth-order TFs shifted subtype fates; and that cellular sex differentiated transcription within subtypes.
Our results demonstrate that cerebral neurons with singular circuit functions are specified through a nested code: hemilineage identity sets the ground plan, whereas birth order carves this plan into distinct subtypes and sexual differentiation overlays additional specialization onto selected cohorts. These separable bits of transcriptional information durably barcode neurons, allowing the brain to build uniquely purposed circuit elements from the same basic developmental logic.
Transcriptional analysis of developing cerebral lineages
Most of the adult D. melanogaster cerebrum derives from approximately 100 spatially patterned, bilaterally symmetric type I neuroblasts, which each divide 70–100 times from embryonic to pupal stages as they progress through expression of temporal TFs and RNA-binding proteins24. Neuroblast asymmetric divisions produce a ganglion mother cell (GMC), and these divide once to produce two neurons, which assume NotchON versus NotchOFF fates6,7,25. The 70–100 neurons of the same neuroblast that share Notch status form a hemilineage. To interrogate hemilineage patterning in the cerebrum, we used an intersectional genetic approach to lineage trace the progeny of identified cerebral neuroblasts that produce well-studied, fruitless-expressing neurons in the male mating circuit (Fig. 1a). In this system, expression of GAL4 within the neuroblast initiates a recombination cascade, resulting in permanent expression of LexA within its progeny (Fig. 1b)5. We drove lineage tracing using the R19C05Pdfr enhancer, which has been empirically shown to be expressed in the CREa1 and SMPad1 neuroblasts, among additional lineages (Fig. 1b,d)22. The NotchOFF (B) hemilineage from CREa1 includes fruitless mAL neurons, which inhibit courtship-promoting P1 neurons in the presence of males or heterospecific females18,26,27,28. The NotchON (A) hemilineage from SMPad1 includes fruitless aSP2 neurons, which are activated in the presence of females and enhance courtship persistence3,4,10,29. We registered twenty labelled brains to a standard template and annotated 13 lineages labelled in at least half of hemispheres, and 13 further clones labelled less frequently (Fig. 1d, Methods and Extended Data Fig. 2). Using this method, all neurons born after the completion of the recombination cascade are labelled, thus embryonically born (primary) neurons may not be included.
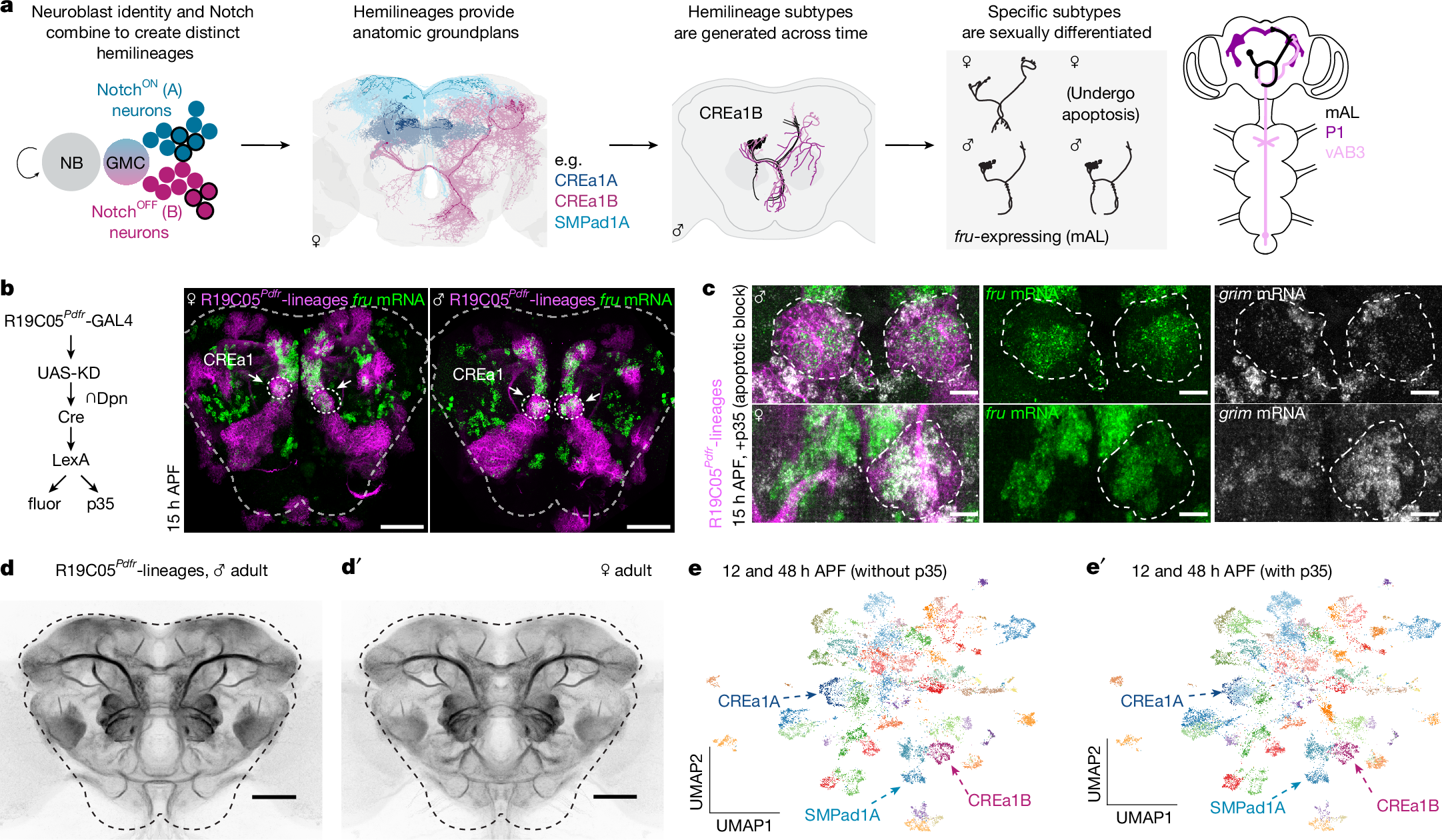
a, Type I neuroblasts generate hemilineages, which form anatomic highways structuring arthropod cerebral circuits. The D. melanogaster CREa1B hemilineage produces fruitless mAL neurons (black) and other morphologies across time. Furthest right: mAL neurons receive gustatory pheromone input from vAB3 and inhibit P1 neurons to gate courtship15,18,26,27. b, Intersection of R19C05Pdfr enhancer with deadpan allows lineage tracing of CREa1 and other neuroblasts (magenta)5,22, including their fruitless progeny (RNA FISH, green). Dashed lines outline the central brain. Further information is provided in Extended Data Fig. 1 and the Methods. Scale bars, 50 μm. c, Clonal induction of p35 in the CREa1 lineage prevents the completion of apoptosis. CREa1B soma marked by R19C05Pdfr-lineage labelling, combined with RNA FISH for fru (green) and grim (grey). grim transcript marks neurons fated to apoptose. In the female sample shown, CREa1B is labelled (and p35 expressed) only in the right hemisphere. In males, fru is translated into FruM and suppresses grim. Scale bars, 10 μm. d,d′, Registered average signal of ten male or female R19C05Pdfr-lineage-traced brains, highlighting hemilineage tract morphologies. Census of labelled lineages provided in Extended Data Fig. 2. Scale bars, 50 μm. e,e′, We performed scRNA-seq transcriptional analysis of developing R19C05Pdfr-lineage neurons (12 h and 48 h APF), with and without the p35 apoptosis block. We clustered neurons using TF transcripts. Quality control and analysis details are provided in Extended Data Fig. 1, whereas CREa1A, CREa1B and SMPad1A hemilineage markers are provided in Extended Data Fig. 2. Allele and full genotype tables are provided in the Methods.
Some neuroblasts produce two living hemilineages with distinct ground plans and neurotransmitter usage, whereas others produce one living hemilineage and one that undergoes programmed cell death in part or in full6,7,18,30. Sexually differentiated cell death is also common26. We therefore genetically blocked the completion of apoptosis by expressing the caspase inhibitor p35 using our lineage labelling strategy (Fig. 1b,c)22. Resurrected cells were easily identified in histology due to their enduring high-level transcription of the pro-apoptotic gene grim (Fig. 1c). Using past clonal analyses, connectomic annotations and comparative histology in brains with and without p35, we confirmed and updated which pairs of living hemilineages are sisters, and which are lone hemilineages, with apoptosing sisters (Methods and Supplementary Table 1).
Having established the ground truth of the lineages included in our genotype, we used fluorescence-activated cell sorting (FACS) of labelled neurons from both sexes, with and without p35, and performed single-cell RNA sequencing (scRNA-seq) at 12 h after puparium formation (APF, around the end of neurogenesis) and 48 h APF (when neurites have reached target neuropils and are beginning to contact potential partners; Extended Data Fig. 1). We retained central brain neurons and sexed them post hoc using expression of the male-specific lncRNAs rox1 and rox2 (Methods and Extended Data Fig. 1). To study the diversification of neuronal fates, we performed semi-supervised clustering using the 628 annotated D. melanogaster TFs. After integrating time points, we tested a variety of methods and resolutions to probe how the four logical axes of diversity related to patterns of TF expression in the dataset, and selected an intermediate resolution on the basis of standard bioinformatic tools, producing 55 clusters after further manual curation (Fig. 1e, Methods and Extended Data Fig.