Ultra-high-throughput mapping of genetic design space
TL;DR
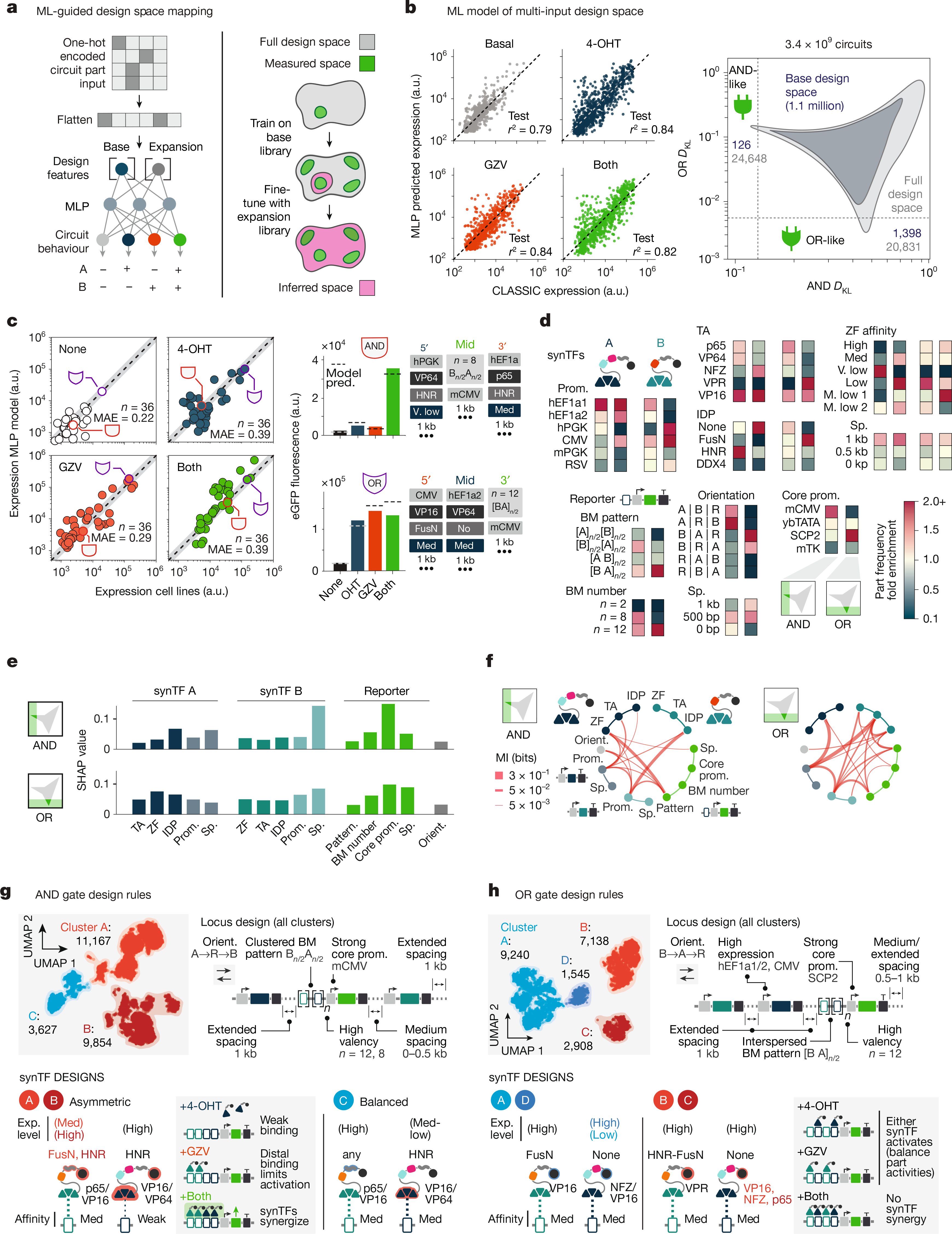
CLASSIC combines long- and short-read sequencing to screen over 100,000 gene circuit designs (5-20 kb) in human cells. It enables machine learning models to predict circuit behavior and reveal part composability rules, accelerating synthetic biology design cycles.
Key Takeaways
- •CLASSIC platform combines long- and short-read NGS to screen complex genetic constructs of arbitrary length
- •Can profile over 100,000 gene circuit designs (5-20 kb) in a single experiment in human cells
- •Enables training of machine learning models that accurately predict circuit behavior across design landscapes
- •Reveals genetic part composability rules that govern circuit performance
- •Accelerates synthetic biology design-build-test-learn cycles for complex genetic systems
Tags
Abstract
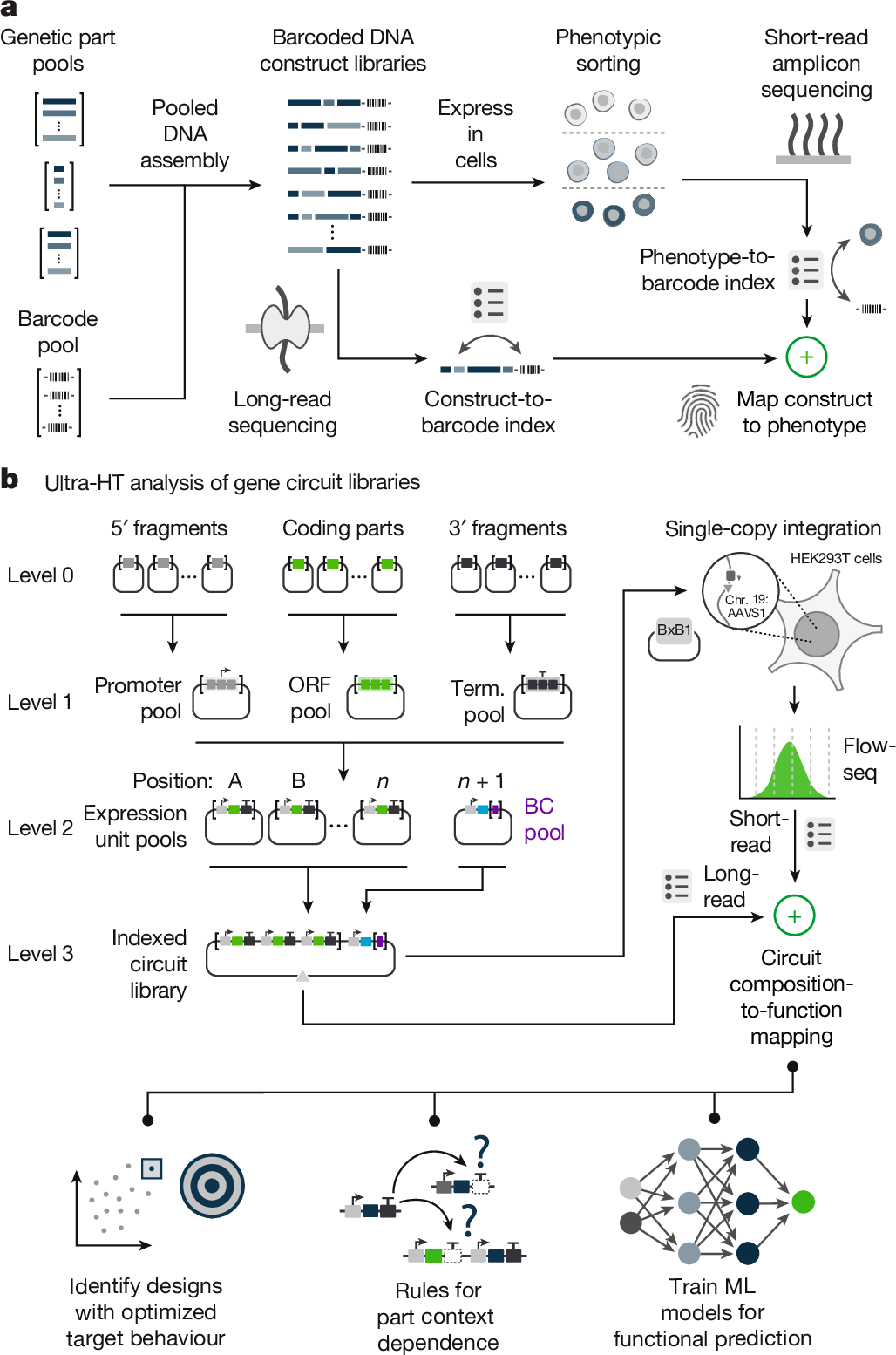
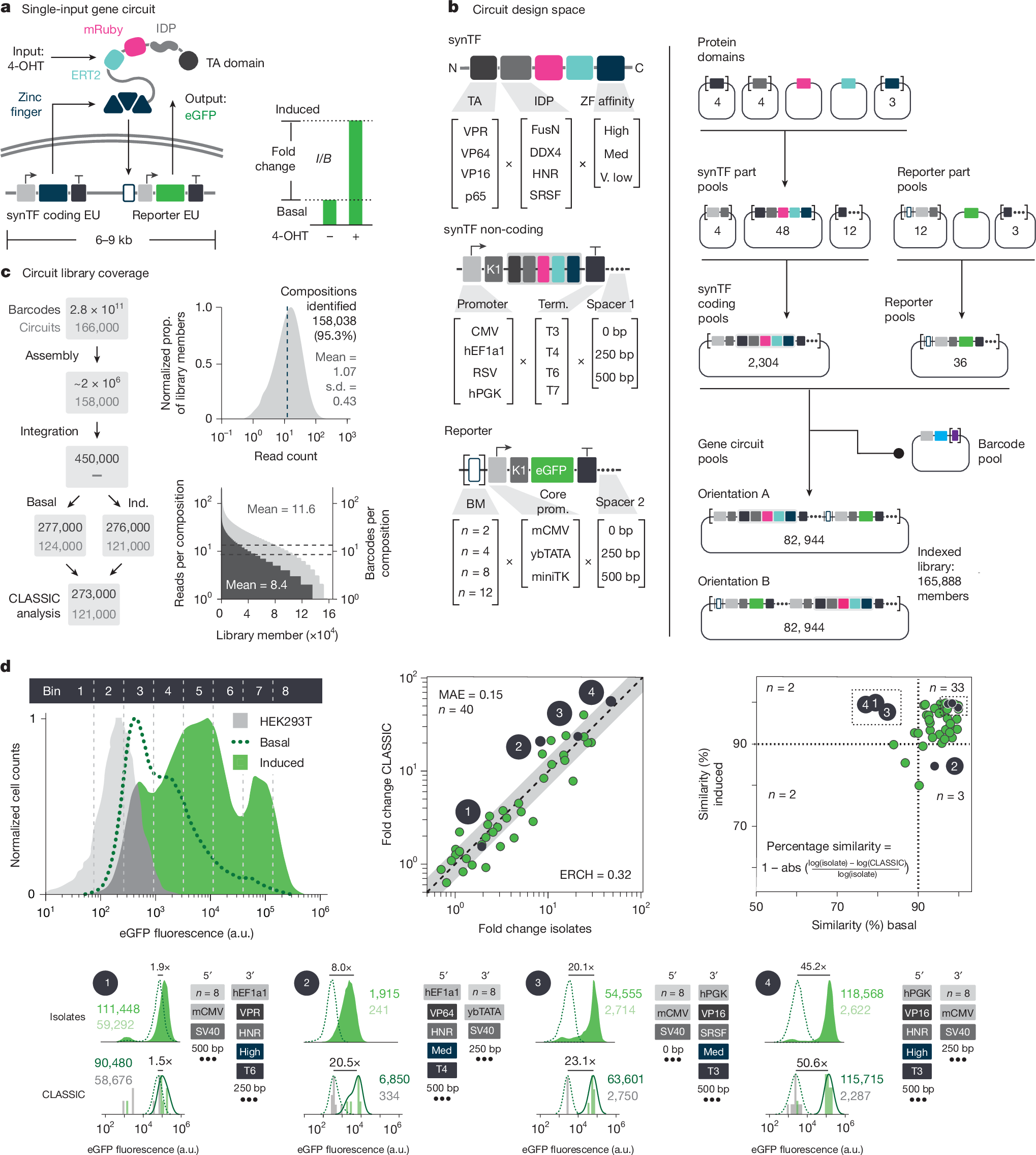
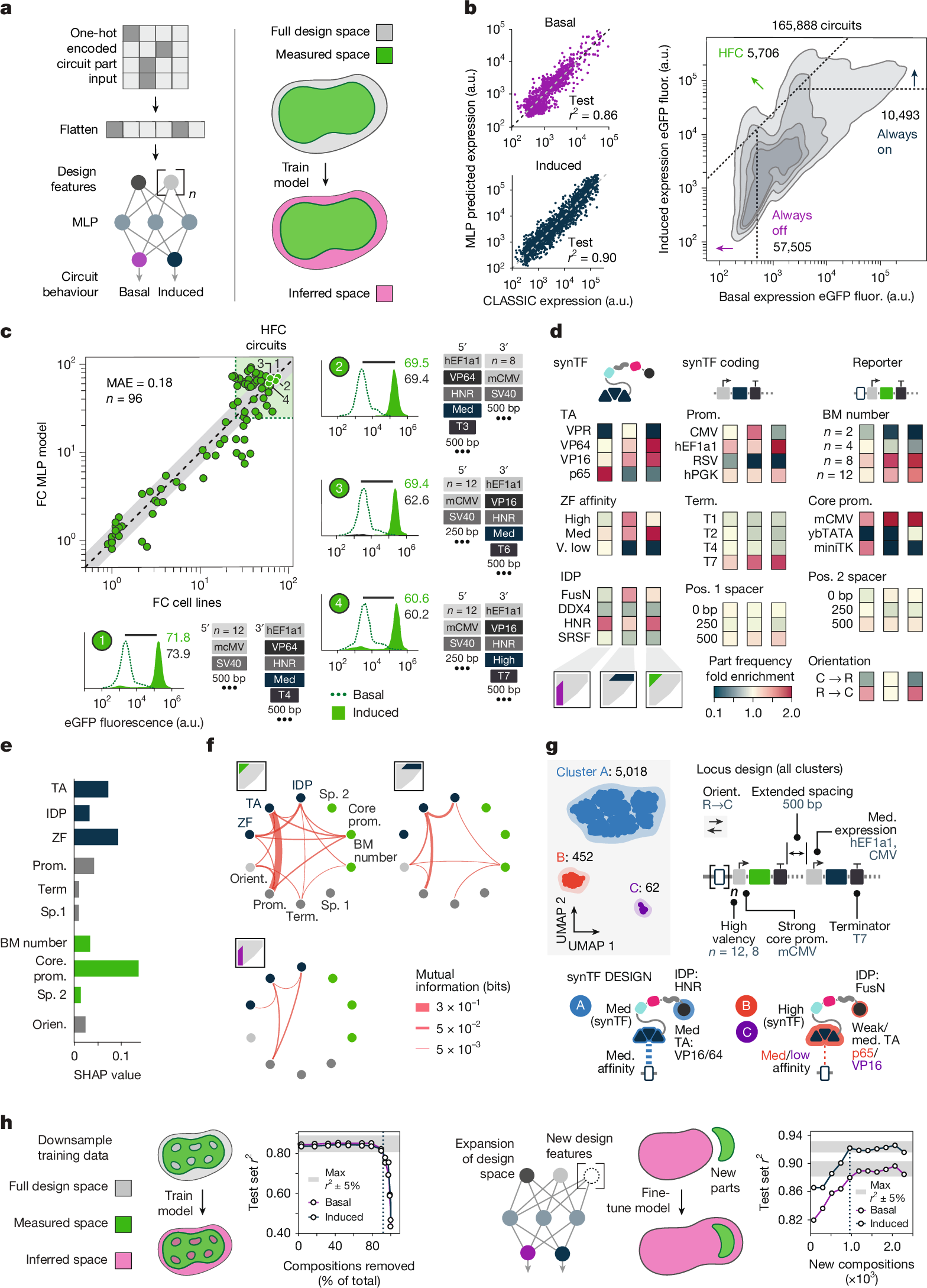
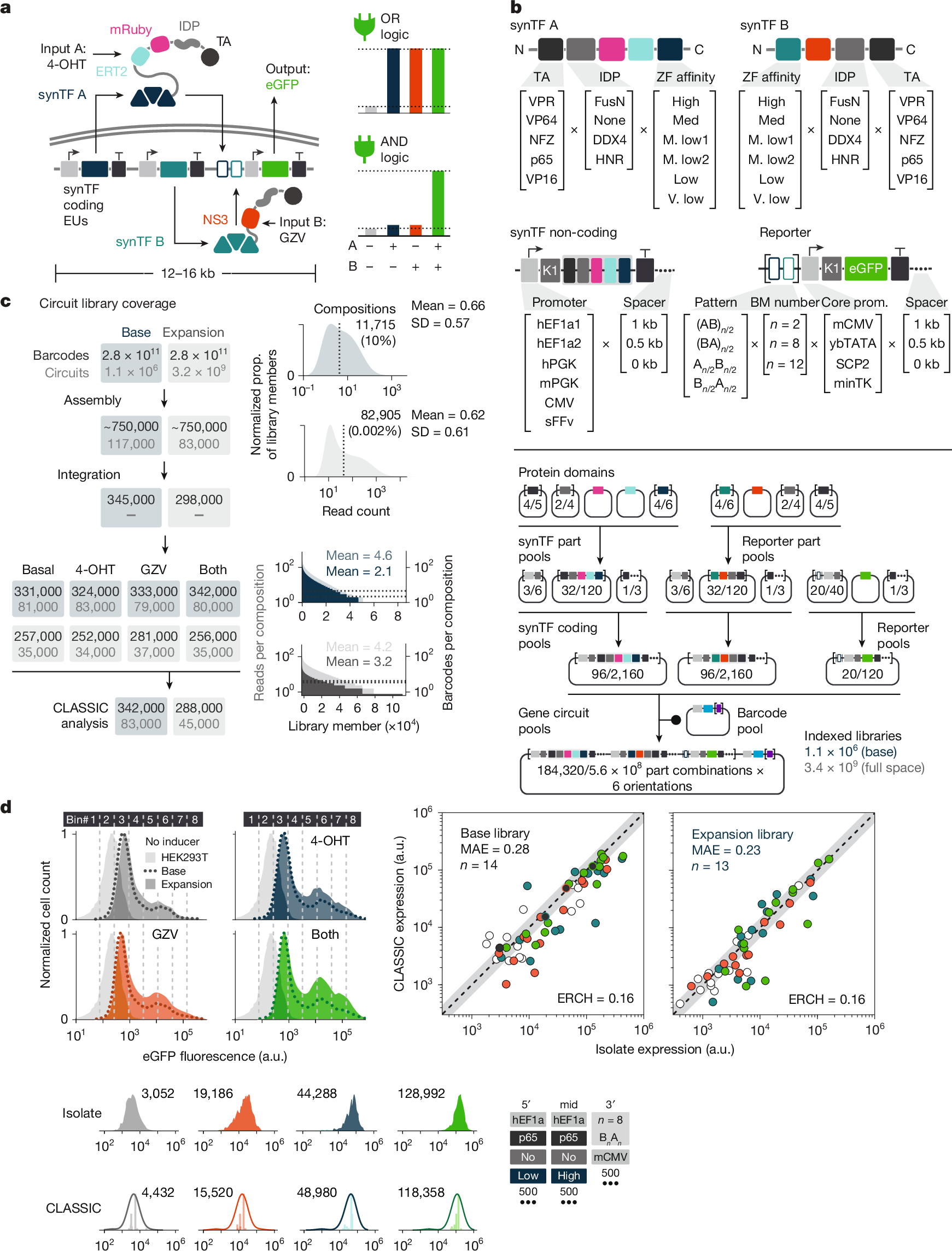
Massively parallel genetic screens have been used to map sequence-to-function relationships for a variety of genetic elements1,2,3,4,5. However, as these approaches interrogate only short sequences, it remains challenging to perform high-throughput assays on constructs containing combinations of multiple sequence elements arranged across multi-kb length scales. Overcoming this barrier could accelerate synthetic biology; by screening diverse gene circuit designs and learning ‘composition to function’ mappings, genetic part composability rules could be revealed, enabling rapid identification of behaviour-optimized design variants6,7. Here we introduce CLASSIC (combining long- and short-range sequencing to investigate genetic complexity), a genetic screening platform that combines long- and short-read next-generation sequencing (NGS) modalities to quantitatively assess pools of constructs of arbitrary length containing diverse genetic part compositions. We show that CLASSIC can measure expression profiles of over 105 gene circuit designs (from 5–20 kb) in a single experiment in human cells. The resulting datasets can be used to train machine-learning models that accurately predict circuit behaviour across expansive circuit design landscapes, revealing part composability rules that govern circuit performance. Our study shows that, by expanding the throughput of each design–build–test–learn cycle, CLASSIC enhances the pace and scale of synthetic biology and establishes an experimental basis for data-driven design of complex genetic systems.
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout
Data availability
All Nanopore and Illumina sequencing datasets generated in this study are available from the Sequencing Read Archive (BioProject: PRJNA1347054).
Code availability
All custom scripts used for Nanopore sequencing data analysis are available at GitHub (https://github.com/cbashorlab/WIMPY). Code associated with Illumina data analysis and model training are available at GitHub (https://github.com/cbashorlab/CLASSIC). All other scripts used to generate any analysis in addition to those provided above are available on request.
References
de Boer, C. G. et al. Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol. 38, 56–65 (2020).
Castillo-Hair, S. et al. Optimizing 5′UTRs for mRNA-delivered gene editing using deep learning. Nat. Commun. 15, 5284 (2024).
Angenent-Mari, N. M., Garruss, A. S., Soenksen, L. R., Church, G. & Collins, J. J. A deep learning approach to programmable RNA switches. Nat. Commun. 11, 5057 (2020).
Sahu, B. et al. Sequence determinants of human gene regulatory elements. Nat. Genet. 54, 283–294 (2022).
Jones, E. M. et al. Structural and functional characterization of G protein-coupled receptors with deep mutational scanning. eLife 9, e54895 (2020).
Zhang, C., Tsoi, R. & You, L. Addressing biological uncertainties in engineering gene circuits. Integr. Biol. 8, 456–464 (2016).
Kitano, S., Lin, C., Foo, J. L. & Chang, M. W. Synthetic biology: learning the way toward high-precision biological design. PLoS Biol. 21, e3002116 (2023).